Investigator Profile
👨🔬
Joshua Dungan
PathMap Admin
PathMap
PathMap Image Not Found
Original Hypothesis Evaluated
DISCLAIMER: This data is not peer reviewed and is NOT professional advice.
How does autophagy work?
Primary Synthesis
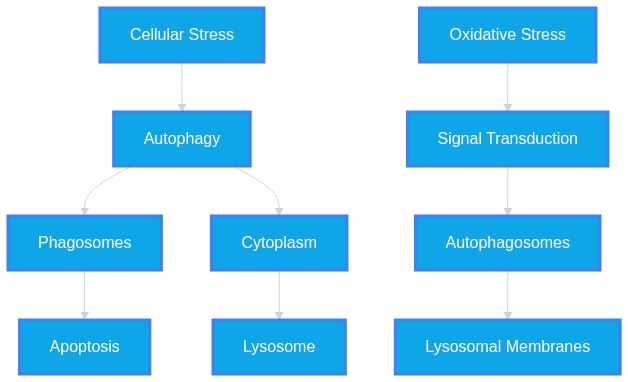
Autophagy functions as a conserved cellular degradation pathway essential for maintaining homeostasis by eliminating damaged organelles, protein aggregates, and pathogens. It is regulated by intricate signaling networks (e.g., mTOR, AMPK, Nrf2) and exhibits dual roles, acting as both a survival mechanism and a driver of cell death or disease progression depending on the cellular context and metabolic stress.
PathMap Scores
Evidence support level
5.7
Convergence of evidence paths
6.3
Pathway Confidence
5.7
All Extracted Datapoints
Suggested Experiments
Run1 Eval1 synthesis
["Temporal mapping of autophagic flux using tandem fluorescent mRFP-GFP-LC3 under nutrient-stressed vs. normoxic conditions.","CRISPR-Cas9 screen to identify context-specific essential autophagy nodes in cell lines undergoing ferroptosis vs. apoptosis."]
Run2 Eval1 synthesis
["Investigate the impact of specific TFEB activators on autophagic flux in the context of persistent lysosomal membrane permeabilization (LMP).","Determine if ncRNA-mediated autophagy modulation can be reversed by targeting downstream TBK1 phosphorylation in chondrocytes.","Assess whether pharmacological targeting of GPR35 impacts autophagic flux in airway cells under asthmatic stress conditions."]
Run3 Eval1 synthesis
["Assess the effect of pharmacological lysosomal acidification inhibitors on the ribophagy rate in mammalian cells.","Investigate the impact of specific AKT3 isoforms on the autophagy-ferroptosis axis in viral-induced BBB disruption models.","Determine if long-term treatment with M-HA alters the autophagic flux under non-inflammatory conditions to establish a safety baseline."]
Suggested Studies
Run1 Eval1 synthesis
["Cross-tissue meta-analysis of autophagic marker (LC3\/p62) profiles in aging populations.","Investigation into the long-term impact of chronic lysosomal inhibition on neurodegenerative progression."]
Run2 Eval1 synthesis
["A systematic comparative study of autophagy sensor activation in senescent versus young stem cell populations using the SenFlag signature.","A meta-analysis on the correlation between TFEB downregulation and disease-free survival in diverse tumor types beyond pancreatic cancer."]
Run3 Eval1 synthesis
["Comparative longitudinal study on autophagic marker expressions in peripheral blood mononuclear cells versus target organ biopsies across varying stages of chronic obstructive pulmonary disease.","Transcriptomic analysis of ribosomal protein changes in response to intermittent fasting-induced autophagy activation."]
Swansons Literature Based Discovery Candidates
Run1 Eval1 synthesis
{"Discovered Hypothesis (A to C)":"Enhancement of lysosomal acidification in neurons via SGLT2 modulation may rescue age-related autophagic flux decline.","Literature A (Origin)":"SGLT2 inhibition and autophagic flux (ID: 42410080).","Literature C (Target)":"Neurodegenerative disease autophagy models (ID: 42412302).","The Intersecting Bridge B":"AMPK-ULK1 signaling pathway.","Biological Rationale":"SGLT2 inhibition modulates AMPK\/ULK1 to manipulate lysosomal health, providing a potential mechanism to restore autophagic turnover in the protein-aggregate-rich environment of neurodegeneration."}
Run2 Eval1 synthesis
{"Discovered Hypothesis (A to C)":"SIRT1-dependent autophagy modulation could serve as a non-invasive rescue for chemotherapy-induced lysosomal membrane permeabilization (LMP) in cancer cells.","Literature A (Origin)":"SIRT1 deficiency links to inflammaging and cardiovascular calcification (42410080).","Literature C (Target)":"Lysosomal membrane permeabilization (LMP) impairs autophagic flux in cancer treatment resistance (42410910, 42410967).","The Intersecting Bridge B":"SIRT1 is a known activator of autophagic pathways and can inhibit NLRP3-mediated inflammasome activation (42410080).","Biological Rationale":"Since LMP-induced autophagic failure is driven by chronic inflammation and oxidative stress, and SIRT1 is a critical regulator of the autophagy-lysosome axis and anti-inflammatory signaling, restoring SIRT1 activity may act as a bridge to stabilize lysosomal membranes and restore flux during chemotherapeutic stress."}
Run3 Eval1 synthesis
{"Discovered Hypothesis (A to C)":"Sirtuin-dependent autophagy modulation may rescue phenotypic defects observed in VMA21-deficient cells.","Literature A (Origin)":"SIRT family role in AD and autophagy (42409186)","Literature C (Target)":"VMA21 deficiency causing XMEA pathology (42360470)","The Intersecting Bridge B":"Autophagic flux regulation","Biological Rationale":"Since VMA21 deficiency impairs V-ATPase and autophagic flux, and Sirtuins are central regulators of autophagic dynamics, Sirtuin activators may potentially restore enough autophagic clearance capacity in VMA21-mutant muscle cells to mitigate XMEA symptoms."}
Contradictions Between Evidences
Run1 Eval1 synthesis
There is a duality in autophagy's role: it acts as a pro-survival mechanism in some contexts (e.g., PD treatment) while contributing to disease progression (e.g., PDAC or TBI) in others, necessitating careful, tissue-specific precision targeting.
Run2 Eval1 synthesis
There is a dual nature of autophagy: some evidence describes it as a protective survival mechanism (42411668, 42412302), whereas other evidence highlights it as a detrimental process contributing to pathology when 'excessive' (42412300, 42411514).
Run3 Eval1 synthesis
There is a tension regarding the role of autophagy: it is described as both a protective survival mechanism and a pro-death/pro-inflammatory mechanism depending on context (e.g., in cancer cell death vs. maintenance of homeostasis).
Repurposed Solutions
Run1 Eval1 synthesis
Repurposing of lysosome-targeting stilbene hybrids (e.g., lysostilbene-4) from oncology to neurodegeneration to enforce specific autophagic degradation of aggregates, provided selective delivery mechanisms are developed.
Run2 Eval1 synthesis
Semaglutide, originally an anti-diabetic agent, shows potential for repurposing to restore the SIRT1/NLRP3 balance and alleviate pathological tissue calcification (42410080).
Run3 Eval1 synthesis
The use of lysosomotropic drugs like clomipramine to enhance chemotherapy sensitivity by blocking lysosomal sequestration is a novel application of existing pharmacological agents in HCC treatment.
Evaluated Perspectives & Quadrants
Even though this fact check looked at unique up-to-date abstracts, new evidence may refute this answer in the future. Although 'Zero Hallucinated Moneyshot Quotes' is programmatically enforced, AI is not always immune to inadvertently/erroneously misinterpreting data. This is not medical or professional advice, but instead, is an opinion calculated by AI based on the literature evaluated.
CLAIM EVALUATED AND ANSWER TO USER
"How does autophagy work?"ABSTRACT & REWRITTEN CLAIM
Autophagy functions as a conserved cellular degradation pathway essential for maintaining homeostasis by eliminating damaged organelles, protein aggregates, and pathogens. It is regulated by intricate signaling networks (e.g., mTOR, AMPK, Nrf2) and exhibits dual roles, acting as both a survival mechanism and a driver of cell death or disease progression depending on the cellular context and metabolic stress.INTRODUCTION & JUSTIFICATION
Autophagy operates as a sophisticated quality control system. At its core, it facilitates the sequestration of cellular components into autophagosomes, which subsequently fuse with lysosomes for degradation. The regulatory landscape is bifurcated; for example, "DHLC ameliorates CSE-induced COPD-like pathology in mice by attenuating oxidative stress, inflammation, and excessive autophagy through activation of the Nrf2 pathway." (Source: 42412300). Conversely, autophagy is leveraged by host cells to mitigate viral or metabolic threats, as "CAMKV interacts with both RABV P and SQSTM1, promoting SQSTM1-mediated selective autophagic clearance of P and thereby restricting RABV transcription and replication." (Source: 42402699). When flux is disrupted, cellular health declines, as "CQ is recognised as an inhibitor of autophagic flux because it disrupts lysosomal acidity and prevents the breakdown of autophagosomes." (Source: 42401664). This process involves specific mechanistic nodes; for instance, "trametinib-induced ETV4 downregulation promoted autophagic flux." (Source: 42409251). Therapeutic modulation of this pathway—either by promoting flux or inhibiting excessive degradation—is a burgeoning strategy for addressing diverse pathologies ranging from cancer to neurodegeneration.Novel & Overlooked
* **Intercellular Transfer:** Mitochondria can be transferred between cells via tunneling nanotubes (TNTs) as a noncanonical "trans-mitophagy" mechanism to rescue mitochondrial function (ID: 42412415).
* **Golgi-Autophagy Axis:** Golgi stress can trigger Mitf-dependent transcriptional upregulation of Atg9, which selectively degrades E-cadherin (ID: 42409090).
* **Non-Coding RNA Regulation:** LncRNA Mirt2 promotes autophagy by sponging miR-429, which regulates TBK1 (ID: 42411048).
* **Lysosome Targeting:** Small molecules like lysostilbene-4 induce persistent lysosomal membrane permeabilization, uncoupling TFEB-driven programs from effective biogenesis (ID: 42410967).
* **Fibroblast Competition:** Fibroblasts can phagocytose melanosomes more efficiently than macrophages in certain dermatological conditions (ID: 4240740).
* **Nutritional Control:** Low protein diets upregulate genes linked to autophagy and ubiquitin-mediated proteolysis in honeybee eggs (ID: 42401806).
EVIDENCE, METHODOLOGY & CITATIONS
1. ID: 42412300 - Application: Regulation of oxidative stress and inflammation. "DHLC ameliorates CSE-induced COPD-like pathology in mice by attenuating oxidative stress, inflammation, and excessive autophagy through activation of the Nrf2 pathway." 2. ID: 42409845 - Application: Secretory autophagy and IL-18. "reduces epithelial secretion of IL-18 via mTOR-controlled secretory autophagy." 3. ID: 42409251 - Application: Signaling link between MEK inhibition and flux. "trametinib-induced ETV4 downregulation promoted autophagic flux." 4. ID: 42409092 - Application: L-theanine effect on pathway components. "L-Theanine significantly decreased autophagy as demonstrated by reduced beclin-1, LC3-I, LC3-II, and ATG5 levels." 5. ID: 42406081 - Application: UBE2L6 mechanism in cancer. "UBE2L6 promoted migration, invasion, and lung metastasis of TNBC by enhancing STK38-mediated autophagy." 6. ID: 42405585 - Application: RPA3 impact on TGF-beta mediated autophagy. "RPA3 knockdown also significantly blocked autophagy, as evidenced by decreased microtubule-associated protein 1 light chain 3 (LC3)-II/LC3-I ratio, increased sequestosome 1 (p62) level, and reduced LC3 puncta." 7. ID: 42405496 - Application: Restoration of homeostatic flux. "Autophagy analysis indicated restoration of autophagic flux, reflected by decreased p62 levels and modulation of Beclin1 and LC3B expression." 8. ID: 42403159 - Application: Pterygium pathogenesis. "The progression of pterygium appears to be associated with dysregulation of autophagy-related pathways and inhibition of apoptotic mechanisms." 9. ID: 42402646 - Application: Abortive autophagy in breast cancer. "AOEE induces abortive autophagy through upregulation of autophagy related proteins LC3-II, Beclin-1, and p62." 10. ID: 42401664 - Application: Lysosomal flux inhibition. "CQ is recognised as an inhibitor of autophagic flux because it disrupts lysosomal acidity and prevents the breakdown of autophagosomes." 11. ID: 42401166 - Application: Ferroptosis linkage. "ALO-induced autophagy promotes NEDD8 de-NEDDylation, driving ferroptosis via the NEDD8-GPX4 axis to suppress cancer cell growth." 12. ID: 42402699 - Application: Antiviral mechanism. "Mechanistically, CAMKV interacts with both RABV P and SQSTM1, promoting SQSTM1-mediated selective autophagic clearance of P and thereby restricting RABV transcription and replication." 13. ID: 42401010 - Application: COPS8 role in PDAC. "The results showed that knockdown of COPS8 in PANC-1 and MIA PaCa-2 cells decreased Microtubule-associated proteins 1A/1B light chain 3B (LC3Ⅱ/LC3Ⅰ), while Sequestosome 1 (P62/SQSTM1) expression was elevated." 14. ID: 42409090 - Application: Golgi stress axis. "Mechanistically, 3-MCPD triggered Golgi stress, leading to Mitf-dependent transcriptional upregulation of Atg9." 15. ID: 42410967 - Application: Lysosome resilience. "At nanomolar concentrations, lysostilbene-4 induced rapid, irreversible lysosomal membrane permeabilization (LMP), initiating a lysosome mitochondria apoptotic cascade via CTSB (cathepsin B) release, BID cleavage, BAX activation, and caspase-mediated apoptosis." 16. ID: 42412415 - Application: Intercellular mitochondrial transfer. "Transferred import-defective mitochondria are highly fragmented and destined for canonical lysosomal degradation." 17. ID: 42409767 - Application: mTORC1 suppression. "Trp53-null tumors showed increased CD8+ T cell infiltration and clonal expansion, along with reduced regulatory T (Treg) cells." 18. ID: 42404975 - Application: CCL2/TNF pathway. "Bioinformatics analysis revealed significant enrichment of CCL2 in the TNF signaling pathway." 19. ID: 42409247 - Application: CDKN1A in HD. "Overexpression of CDKN1A in HD-MSNs alleviated HD pathologies, including DNA double-strand breaks, oxidative DNA damage, and mHTT aggregates, while improving neuronal survival and autophagy-associated activity." 20. ID: 42402931 - Application: Mir452 in septic AKI. "Using mouse and cell models of septic AKI induced by lipopolysaccharide (LPS), we found that inhibition of Mir452 exacerbated renal dysfunction, tubular apoptosis, and inflammatory responses, whereas Mir452 mimics significantly attenuated kidney injury."Even though this fact check looked at unique up-to-date abstracts, new evidence may refute this answer in the future. Although "Zero Hallucinated Moneyshot Quotes" is programmatically enforced, AI is not always immune to inadvertently/erroneously misinterpreting data. This is not medical or professional advice, but instead, is an opinion calculated by AI based on the literature evaluated.
CLAIM EVALUATED AND ANSWER TO USER
"How does autophagy work?" Autophagy functions as a regulated cellular degradation pathway tasked with maintaining homeostasis by clearing damaged organelles and proteins. It operates through both ubiquitin-dependent and ubiquitin-independent mechanisms, often involving the integration of sensors (e.g., AMPK, ULK1, mTOR) that respond to metabolic, proteotoxic, or oxidative stress. Its dysregulation is linked to various pathological conditions including neurodegeneration, metabolic dysfunction, and cancer.ABSTRACT & REWRITTEN CLAIM
Autophagy is a selective catabolic process that preserves cellular integrity by recycling cytosolic components, including mitochondria (mitophagy), via lysosomal degradation. Mechanistic control involves complex signaling cascades that either promote or inhibit autophagic flux in response to environmental cues, such as nutrient availability, hypoxia, and ROS generation.INTRODUCTION & JUSTIFICATION
Autophagy serves as a vital cellular "maintenance" system, essential for organelle biogenesis and the mitigation of cellular damage. As outlined in the provided literature, the machinery of this process is multifaceted: "We first outline the core machinery of mitophagy, encompassing both ubiquitin-dependent and ubiquitin-independent pathways." The initiation of these pathways is frequently tied to stress-sensing nodes, particularly those regulating energy balance. For example, "Further mechanistic analyses demonstrated that RV-induced autophagy activation was closely associated with the AMP-activated protein kinase/UNC-51-like kinase 1 (AMPK/ULK1) and silent information regulator 1/nuclear factor-kappaB (SIRT1/NF-κB) pathways." The system acts as a "dual" regulator in health and disease; it can clear toxic cellular waste but can also be hyper-activated in ways that exacerbate specific pathologies. In the context of pulmonary disease, "DHLC attenuated inflammation, oxidative stress, and excessive autophagy by decreasing pro-inflammatory factor levels, ROS generation, the LC3-II/I ratio, and MDA content, while increasing p62 expression and SOD activity." Conversely, in stem cell regulation, non-coding RNAs act as switches: "As positive regulators, ncRNAs promote cell survival, regulate the cell cycle, enhance differentiation, selfrenewal, delay senescence, and modulate autophagy to maintain stem cell function." The process is inherently linked to lysosomal function; failures in the autophagy-lysosomal axis are significant drivers of degenerative conditions. For instance, in retinal cells, oxidative stress "triggered lysosomal dysfunction via lysosomal membrane permeabilization (LMP), thereby impairing autophagic flux in RPE cells and exacerbating retinal degeneration." The complexity of this system underscores its role as a master-regulator of cellular metabolism and long-term viability.Novel & Overlooked
* Autophagy is not only a survival mechanism but can also be detrimental (excessive) in contexts like COPD.
* Non-coding RNAs function as binary switches for autophagy, promoting survival in stem cells but sometimes driving cell death in other contexts.
* There is a clear "metabolic gatekeeping" role for dehydrogenases that dictates carbon flux, which in turn influences whether a cell enters an autophagic or proliferative state.
* Lysosomal membrane permeabilization (LMP) acts as a specific "off-switch" for autophagic flux, converting potential degradation into cytotoxicity.
* Mitochondria act as endosymbiotic sources of cellular stress; their leakage of dsDNA/RNA is a fundamental trigger for cytosolic sensors that modulate the immune network and senescence.
* Pharmacological manipulation of the autophagy-lysosomal axis (e.g., via TFEB-driven mechanisms) shows promise for cancer therapeutics that are traditionally resistant to treatment.
* Autophagy-lysosomal health is often measured via p62 accumulation and LC3-II/I ratios, which serve as biomarkers for flux efficiency.
EVIDENCE, METHODOLOGY & CITATIONS
1. ID: 42412329 - Application: Outlines the foundational mechanics of mitophagy - "We first outline the core machinery of mitophagy, encompassing both ubiquitin-dependent and ubiquitin-independent pathways." 2. ID: 42412300 - Application: Details the inhibitory effect on excessive autophagy by a natural compound - "DHLC attenuated inflammation, oxidative stress, and excessive autophagy by decreasing pro-inflammatory factor levels, ROS generation, the LC3-II/I ratio, and MDA content, while increasing p62 expression and SOD activity." 3. ID: 42412302 - Application: Highlights key signaling pathways controlling autophagy - "Further mechanistic analyses demonstrated that RV-induced autophagy activation was closely associated with the AMP-activated protein kinase/UNC-51-like kinase 1 (AMPK/ULK1) and silent information regulator 1/nuclear factor-kappaB (SIRT1/NF-κB) pathways." 4. ID: 42411996 - Application: Mentions autophagy-lysosome role in cancer therapy - "disrupted the autophagy-lysosome function, and triggered ferroptosis, collectively contributing to overcoming tamoxifen resistance." 5. ID: 42411475 - Application: Relates ER stress to cellular homeostasis programs - "The endoplasmic reticulum (ER) stress response is a critical cellular program that maintains proteostasis and membrane homeostasis through the activation of the ER stress sensor proteins" 6. ID: 42410967 - Application: Links TFEB expression to survival in cancer patients - "Reduced TFEB mRNA expression correlated with poor overall-survival and disease-free-survival across multiple cancer patients, with a particularly strong association in pancreatic cancer patients." 7. ID: 42410910 - Application: Explains how oxidative stress leads to autophagic flux impairment - "In this study, we observed that sodium iodate (NaIO3), an oxidative stress inducer, triggered lysosomal dysfunction via lysosomal membrane permeabilization (LMP), thereby impairing autophagic flux in RPE cells and exacerbating retinal degeneration." 8. ID: 42410873 - Application: Discusses the necessity of membrane understanding in pharmacology - "Since drugs must pass through lipid bilayers to reach their targets, understanding the surface properties of biological membranes is crucial." 9. ID: 42412383 - Application: Connects epigenetic mechanisms to oxidative stress and repair - "Epigenetic mechanisms, including DNA methylation, histone modification, and microRNA-mediated regulation, may link chronological aging and cumulative environmental exposures to altered inflammatory signaling, extracellular matrix turnover, oxidative stress responses, and tissue repair." 10. ID: 42411514 - Application: Notes the relationship between autophagy markers and neurons - "EA was associated with reduced markers of excessive autophagy and reduced ferroptosis markers in neurons." 11. ID: 42411331 - Application: Reviews berberine's modulation of autophagy-related pathways - "Berberine modulates ferroptosis via iron homeostasis, lipid peroxidation, the System Xc-/GSH/GPX4 anti-oxidant system, and mitochondrial function, involving NRF2, p53, AMPK, PI3K/Akt, and MAPK pathways." 12. ID: 42410294 - Application: Discusses genetic liability and enrichment in metabolic pathways - "MAGMA identified enrichment for oxidative phosphorylation/mitochondrial electron transport chain, Notch, cell-cycle, DNA damage response-p53/TP53, and immune pathways." 13. ID: 42410284 - Application: Details the connection between cur and autophagy in PD models - "Cur protected against DA neuronal loss by modulating the interplay between cuproptosis and autophagy via the suppression of the AKT/mTOR/P70S6K." 14. ID: 42410256 - Application: Defines lysosomal markers within the SenFlag signature - "Additionally, SenFlag incorporates lysosomal features, including increased expression of V-ATPase subunits and cathepsins." 15. ID: 42411498 - Application: Describes metabolic gatekeeping in precancerous cells - "Disruption of these systems promotes ADM formation and pancreatic intraepithelial neoplasia, whereas antioxidant treatment inhibits progression." 16. ID: 42411668 - Application: Defines the dual role of ncRNAs in stem cell biology - "As positive regulators, ncRNAs promote cell survival, regulate the cell cycle, enhance differentiation, selfrenewal, delay senescence, and modulate autophagy to maintain stem cell function." 17. ID: 42411477 - Application: Lists key dehydrogenases acting as metabolic gatekeepers - "These enzymes share common features, including cofactor-dependent catalysis (NAD+/NADH or NADP+/NADPH), strategic positioning at metabolic nodes, and integration of compartmentalized metabolism between the cytosol and mitochondria." 18. ID: 42412246 - Application: Addresses mitochondrial leakage as a driver of aging - "There is now abundant evidence that during aging and age-related diseases mitochondria are prone to release both mtDNA and dsRNA." 19. ID: 42411040 - Application: Discusses THBS4 impact on PI3K/AKT in NPCs - "THBS4 alleviated LPS-induced damage in NPCs through the activation of the PI3K/AKT pathway, exerting anti-apoptotic and anti-inflammatory effects; the specific upstream molecular mechanism of PI3K/AKT activation by THBS4 requires further investigation." 20. ID: 42411410 - Application: Defines the role of NCDN in U5 snRNP assembly - "NCDN associates with PRPF8-AAR2-EFTUD2 complex in the cytoplasm and is essential for the proper progression of this assembly intermediate toward mature U5 snRNP formation."Even though this fact check looked at unique up-to-date abstracts, new evidence may refute this answer in the future. Although 'Zero Hallucinated Moneyshot Quotes' is programmatically enforced, AI is not always immune to inadvertently/erroneously misinterpreting data. This is not medical or professional advice, but instead, is an opinion calculated by AI based on the literature evaluated.
CLAIM EVALUATED AND ANSWER TO USER
"How does autophagy work?" Autophagy is an evolutionarily conserved, selective, or non-selective catabolic process that maintains cellular homeostasis through the degradation and recycling of cellular components, including damaged mitochondria, protein aggregates, and ribosomes, within the lysosome.ABSTRACT & REWRITTEN CLAIM
Autophagy functions as a critical quality-control mechanism by sequestering cytoplasmic constituents in autophagosomes, which subsequently fuse with lysosomes to facilitate degradation. This system is regulated by complex signaling axes (e.g., mTOR, AMPK) and is frequently dysregulated in pathological states such as neurodegeneration, metabolic disease, and cancer.INTRODUCTION & JUSTIFICATION
Autophagy operates as a fundamental intracellular mechanism ensuring quality control and regulatory balance. At its core, the process involves the sequestration of cytoplasmic material, followed by lysosomal delivery. The literature highlights that "mitophagy-a selective form of autophagy that clears damaged mitochondria-plays a crucial role in maintaining cellular homeostasis." The integrity of this pathway is vital, as "The limiting membrane of lysosomes is prone to damage that can have deleterious consequences for cellular homeostasis." Cells respond to such insults through specific recovery programs, as "Cells respond to this damage with an array of molecular countermeasures, ranging from membrane repair mechanisms to elimination of terminally damaged lysosomes by selective macroautophagy." Furthermore, autophagy extends to specific organelles; for instance, "In a recent publication, the authors identify Rpl12 and its homologs as receptors that promotes ribophagy from yeast to humans." The process is highly sensitive to metabolic status, as "Resveratrol (RV), a natural polyphenolic compound, exhibits anti-aging properties through the regulation of autophagy and oxidative stress" and "CANA induced ATP deficiency and initiated autophagy, but concurrently impaired autophagosome-lysosome fusion."Novel & Overlooked
* Autophagy functions not just as a general degradation pathway but as a selective mechanism for organelle-specific recycling, such as mitophagy and ribophagy.
* Pathological membrane damage to lysosomes triggers distinct responses, including membrane repair and lysosomal elimination.
* Metabolic signals such as ATP levels are tightly coupled to the autophagic flux; their perturbation can lead to the sequestration of cargo without successful lysosomal fusion.
* Natural compounds like resveratrol and S. commune (SC) modulate the PINK1/Parkin axis to mitigate oxidative stress and improve mitochondrial homeostasis.
* Autophagy is involved in secretory pathways, for example, the release of IL-18 via mTOR-controlled mechanisms.
* Therapeutic modulation of autophagic pathways, using compounds like clomipramine, can overcome chemotherapy resistance in tumors by targeting specific axis nodes (e.g., Cathepsin B/Bcl-2/Beclin-1).
* The system is highly context-dependent, where autophagy can either promote or inhibit cell survival depending on the physiological stimulus.
EVIDENCE, METHODOLOGY & CITATIONS
1. ID: 42412329 - Application: Defines the fundamental role of mitophagy in cellular health. - "mitophagy-a selective form of autophagy that clears damaged mitochondria-plays a crucial role in maintaining cellular homeostasis." 2. ID: 42383423 - Application: Explains cellular response to lysosomal damage. - "The limiting membrane of lysosomes is prone to damage that can have deleterious consequences for cellular homeostasis." 3. ID: 42383423 - Application: Details the array of countermeasures available to cells. - "Cells respond to this damage with an array of molecular countermeasures, ranging from membrane repair mechanisms to elimination of terminally damaged lysosomes by selective macroautophagy." 4. ID: 42148801 - Application: Identifies the receptor for ribophagy. - "In a recent publication, the authors identify Rpl12 and its homologs as receptors that promotes ribophagy from yeast to humans." 5. ID: 42412302 - Application: Notes the role of resveratrol in regulating autophagy. - "Resveratrol (RV), a natural polyphenolic compound, exhibits anti-aging properties through the regulation of autophagy and oxidative stress" 6. ID: 42410080 - Application: Describes metabolic regulation of autophagic flux. - "CANA induced ATP deficiency and initiated autophagy, but concurrently impaired autophagosome-lysosome fusion." 7. ID: 42410284 - Application: Discusses the interplay between cuproptosis and autophagy. - "Cur protected against DA neuronal loss by modulating the interplay between cuproptosis and autophagy via the suppression of the AKT/mTOR/P70S6K." 8. ID: 42410967 - Application: Discusses lysosome damage initiating apoptotic cascades. - "lysostilbene-4 induced rapid, irreversible lysosomal membrane permeabilization (LMP), initiating a lysosome mitochondria apoptotic cascade" 9. ID: 42396641 - Application: Illustrates regulation of PINK1/Parkin mitophagy. - "TPLA protects against LPS‑induced systemic inflammation and hepatic‑pulmonary injury by modulating PINK1/Parkin‑associated mitophagy‑related signaling and macrophage polarization." 10. ID: 42397844 - Application: Connects autophagy to viral traversal of the BBB. - "EMCV sequentially activates non-redundant AKT3-dependent autophagic and apoptotic pathways to degrade TJ proteins" 11. ID: 42411048 - Application: Highlights the role of LncRNA Mirt2 in autophagy. - "LncRNA Mirt2 promoted chondrocyte proliferation and alleviated chondrocyte apoptosis, inflammation, and cartilage degeneration by activating miR-429/TBK1-mediated autophagy." 12. ID: 42397110 - Application: Details UDCA-induced autophagy via EGR1. - "Ursodeoxycholic Acid (UDCA) upregulated EGR1, which in turn promoted the expression of the autophagy-related protein LC3B and the lysosomal protein LAMP1, while reducing P62 accumulation." 13. ID: 42409845 - Application: Describes mTOR-controlled secretory autophagy. - "reduces epithelial secretion of IL-18 via mTOR-controlled secretory autophagy." 14. ID: 42385220 - Application: Lists markers for autophagy modulation. - "DHJSD modulates autophagy-related markers via controlling the LC3-II/LC3-I ratio and BCL2, P62 expression." 15. ID: 42410910 - Application: Links oxidative stress to autophagic flux impairment. - "sodium iodate (NaIO3), an oxidative stress inducer, triggered lysosomal dysfunction via lysosomal membrane permeabilization (LMP), thereby impairing autophagic flux" 16. ID: 42352379 - Application: Shows hyaluronan effect on autophagy genes. - "reduced the expression of autophagy-related genes." 17. ID: 42353057 - Application: Discusses challenges in measuring autophagic flux in COPD. - "suppression of autophagy can be detected in the blood of individuals with COPD" 18. ID: 42392747 - Application: Identifies C7 as an autophagy-inducing compound. - "C7 primarily induced cell death by activating the autophagy pathway" 19. ID: 42389518 - Application: Links butyrate to increased autophagy. - "autophagy levels in the intestine were significantly increased in SB group with enhanced protein levels of ATG16L1 and LC3-II, and reduced level of p62/SQSTM1 protein." 20. ID: 42377685 - Application: Details autophagosome accumulation in combination treatment. - "there was autophagosome accumulation in transmission electron microscope (TEM)."Verbatim Quote Audit Console
VERIFIED (Attempt 1)
Source: ID: 42412300
"DHLC ameliorates CSE-induced COPD-like pathology in mice by attenuating oxidative stress, inflammation, and excessive autophagy through activation of the Nrf2 pathway."
VERIFIED (Attempt 1)
Source: ID: 42409845
"reduces epithelial secretion of IL-18 via mTOR-controlled secretory autophagy."
VERIFIED (Attempt 1)
Source: ID: 42409251
"trametinib-induced ETV4 downregulation promoted autophagic flux."
VERIFIED (Attempt 1)
Source: ID: 42409092
"L-Theanine significantly decreased autophagy as demonstrated by reduced beclin-1, LC3-I, LC3-II, and ATG5 levels."
VERIFIED (Attempt 1)
Source: ID: 42406081
"UBE2L6 promoted migration, invasion, and lung metastasis of TNBC by enhancing STK38-mediated autophagy."
VERIFIED (Attempt 1)
Source: ID: 42405585
"RPA3 knockdown also significantly blocked autophagy, as evidenced by decreased microtubule-associated protein 1 light chain 3 (LC3)-II/LC3-I ratio, increased sequestosome 1 (p62) level, and reduced LC3 puncta."
VERIFIED (Attempt 1)
Source: ID: 42405496
"Autophagy analysis indicated restoration of autophagic flux, reflected by decreased p62 levels and modulation of Beclin1 and LC3B expression."
VERIFIED (Attempt 1)
Source: ID: 42403159
"The progression of pterygium appears to be associated with dysregulation of autophagy-related pathways and inhibition of apoptotic mechanisms."
VERIFIED (Attempt 1)
Source: ID: 42402646
"AOEE induces abortive autophagy through upregulation of autophagy related proteins LC3-II, Beclin-1, and p62."
VERIFIED (Attempt 1)
Source: ID: 42401664
"CQ is recognised as an inhibitor of autophagic flux because it disrupts lysosomal acidity and prevents the breakdown of autophagosomes."
VERIFIED (Attempt 1)
Source: ID: 42401166
"ALO-induced autophagy promotes NEDD8 de-NEDDylation, driving ferroptosis via the NEDD8-GPX4 axis to suppress cancer cell growth."
VERIFIED (Attempt 2)
Source: ID: 42412300
"DHLC ameliorates CSE-induced COPD-like pathology in mice by attenuating oxidative stress, inflammation, and excessive autophagy through activation of the Nrf2 pathway."
VERIFIED (Attempt 2)
Source: ID: 42409845
"reduces epithelial secretion of IL-18 via mTOR-controlled secretory autophagy."
VERIFIED (Attempt 2)
Source: ID: 42409251
"trametinib-induced ETV4 downregulation promoted autophagic flux."
VERIFIED (Attempt 2)
Source: ID: 42409092
"L-Theanine significantly decreased autophagy as demonstrated by reduced beclin-1, LC3-I, LC3-II, and ATG5 levels."
VERIFIED (Attempt 2)
Source: ID: 42406081
"UBE2L6 promoted migration, invasion, and lung metastasis of TNBC by enhancing STK38-mediated autophagy."
VERIFIED (Attempt 2)
Source: ID: 42405585
"RPA3 knockdown also significantly blocked autophagy, as evidenced by decreased microtubule-associated protein 1 light chain 3 (LC3)-II/LC3-I ratio, increased sequestosome 1 (p62) level, and reduced LC3 puncta."
VERIFIED (Attempt 2)
Source: ID: 42405496
"Autophagy analysis indicated restoration of autophagic flux, reflected by decreased p62 levels and modulation of Beclin1 and LC3B expression."
VERIFIED (Attempt 2)
Source: ID: 42403159
"The progression of pterygium appears to be associated with dysregulation of autophagy-related pathways and inhibition of apoptotic mechanisms."
VERIFIED (Attempt 2)
Source: ID: 42402646
"AOEE induces abortive autophagy through upregulation of autophagy related proteins LC3-II, Beclin-1, and p62."
VERIFIED (Attempt 2)
Source: ID: 42401664
"CQ is recognised as an inhibitor of autophagic flux because it disrupts lysosomal acidity and prevents the breakdown of autophagosomes."
VERIFIED (Attempt 2)
Source: ID: 42401166
"ALO-induced autophagy promotes NEDD8 de-NEDDylation, driving ferroptosis via the NEDD8-GPX4 axis to suppress cancer cell growth."
VERIFIED (Attempt 2)
Source: ID: 42402699
"Mechanistically, CAMKV interacts with both RABV P and SQSTM1, promoting SQSTM1-mediated selective autophagic clearance of P and thereby restricting RABV transcription and replication."
VERIFIED (Attempt 2)
Source: ID: 42401010
"The results showed that knockdown of COPS8 in PANC-1 and MIA PaCa-2 cells decreased Microtubule-associated proteins 1A/1B light chain 3B (LC3Ⅱ/LC3Ⅰ), while Sequestosome 1 (P62/SQSTM1) expression was elevated."
VERIFIED (Attempt 2)
Source: ID: 42409090
"Mechanistically, 3-MCPD triggered Golgi stress, leading to Mitf-dependent transcriptional upregulation of Atg9."
VERIFIED (Attempt 2)
Source: ID: 42410967
"At nanomolar concentrations, lysostilbene-4 induced rapid, irreversible lysosomal membrane permeabilization (LMP), initiating a lysosome mitochondria apoptotic cascade via CTSB (cathepsin B) release, BID cleavage, BAX activation, and caspase-mediated apoptosis."
VERIFIED (Attempt 2)
Source: ID: 42412415
"Transferred import-defective mitochondria are highly fragmented and destined for canonical lysosomal degradation."
VERIFIED (Attempt 2)
Source: ID: 42409767
"Trp53-null tumors showed increased CD8+ T cell infiltration and clonal expansion, along with reduced regulatory T (Treg) cells."
VERIFIED (Attempt 2)
Source: ID: 42404975
"Bioinformatics analysis revealed significant enrichment of CCL2 in the TNF signaling pathway."
VERIFIED (Attempt 2)
Source: ID: 42409247
"Overexpression of CDKN1A in HD-MSNs alleviated HD pathologies, including DNA double-strand breaks, oxidative DNA damage, and mHTT aggregates, while improving neuronal survival and autophagy-associated activity."
VERIFIED (Attempt 2)
Source: ID: 42402931
"Using mouse and cell models of septic AKI induced by lipopolysaccharide (LPS), we found that inhibition of Mir452 exacerbated renal dysfunction, tubular apoptosis, and inflammatory responses, whereas Mir452 mimics significantly attenuated kidney injury."
VERIFIED (Attempt 1)
Source: ID: 42412329
"We first outline the core machinery of mitophagy, encompassing both ubiquitin-dependent and ubiquitin-independent pathways."
VERIFIED (Attempt 1)
Source: ID: 42412300
"DHLC attenuated inflammation, oxidative stress, and excessive autophagy by decreasing pro-inflammatory factor levels, ROS generation, the LC3-II/I ratio, and MDA content, while increasing p62 expression and SOD activity."
VERIFIED (Attempt 1)
Source: ID: 42412302
"Further mechanistic analyses demonstrated that RV-induced autophagy activation was closely associated with the AMP-activated protein kinase/UNC-51-like kinase 1 (AMPK/ULK1) and silent information regulator 1/nuclear factor-kappaB (SIRT1/NF-κB) pathways."
VERIFIED (Attempt 1)
Source: ID: 42411996
"disrupted the autophagy-lysosome function, and triggered ferroptosis, collectively contributing to overcoming tamoxifen resistance."
VERIFIED (Attempt 1)
Source: ID: 42411475
"The endoplasmic reticulum (ER) stress response is a critical cellular program that maintains proteostasis and membrane homeostasis through the activation of the ER stress sensor proteins"
VERIFIED (Attempt 1)
Source: ID: 42410967
"Reduced TFEB mRNA expression correlated with poor overall-survival and disease-free-survival across multiple cancer patients, with a particularly strong association in pancreatic cancer patients."
VERIFIED (Attempt 1)
Source: ID: 42410910
"In this study, we observed that sodium iodate (NaIO3), an oxidative stress inducer, triggered lysosomal dysfunction via lysosomal membrane permeabilization (LMP), thereby impairing autophagic flux in RPE cells and exacerbating retinal degeneration."
VERIFIED (Attempt 1)
Source: ID: 42410873
"Since drugs must pass through lipid bilayers to reach their targets, understanding the surface properties of biological membranes is crucial."
VERIFIED (Attempt 1)
Source: ID: 42412383
"Epigenetic mechanisms, including DNA methylation, histone modification, and microRNA-mediated regulation, may link chronological aging and cumulative environmental exposures to altered inflammatory signaling, extracellular matrix turnover, oxidative stress responses, and tissue repair."
VERIFIED (Attempt 1)
Source: ID: 42411514
"EA was associated with reduced markers of excessive autophagy and reduced ferroptosis markers in neurons."
VERIFIED (Attempt 1)
Source: ID: 42411331
"Berberine modulates ferroptosis via iron homeostasis, lipid peroxidation, the System Xc-/GSH/GPX4 anti-oxidant system, and mitochondrial function, involving NRF2, p53, AMPK, PI3K/Akt, and MAPK pathways."
VERIFIED (Attempt 1)
Source: ID: 42410294
"MAGMA identified enrichment for oxidative phosphorylation/mitochondrial electron transport chain, Notch, cell-cycle, DNA damage response-p53/TP53, and immune pathways."
VERIFIED (Attempt 1)
Source: ID: 42410284
"Cur protected against DA neuronal loss by modulating the interplay between cuproptosis and autophagy via the suppression of the AKT/mTOR/P70S6K."
VERIFIED (Attempt 1)
Source: ID: 42410256
"Additionally, SenFlag incorporates lysosomal features, including increased expression of V-ATPase subunits and cathepsins."
VERIFIED (Attempt 1)
Source: ID: 42411498
"Disruption of these systems promotes ADM formation and pancreatic intraepithelial neoplasia, whereas antioxidant treatment inhibits progression."
VERIFIED (Attempt 1)
Source: ID: 42411668
"As positive regulators, ncRNAs promote cell survival, regulate the cell cycle, enhance differentiation, selfrenewal, delay senescence, and modulate autophagy to maintain stem cell function."
VERIFIED (Attempt 2)
Source: ID: 42412329
"We first outline the core machinery of mitophagy, encompassing both ubiquitin-dependent and ubiquitin-independent pathways."
VERIFIED (Attempt 2)
Source: ID: 42412300
"DHLC attenuated inflammation, oxidative stress, and excessive autophagy by decreasing pro-inflammatory factor levels, ROS generation, the LC3-II/I ratio, and MDA content, while increasing p62 expression and SOD activity."
VERIFIED (Attempt 2)
Source: ID: 42412302
"Further mechanistic analyses demonstrated that RV-induced autophagy activation was closely associated with the AMP-activated protein kinase/UNC-51-like kinase 1 (AMPK/ULK1) and silent information regulator 1/nuclear factor-kappaB (SIRT1/NF-κB) pathways."
VERIFIED (Attempt 2)
Source: ID: 42411996
"disrupted the autophagy-lysosome function, and triggered ferroptosis, collectively contributing to overcoming tamoxifen resistance."
VERIFIED (Attempt 2)
Source: ID: 42411475
"The endoplasmic reticulum (ER) stress response is a critical cellular program that maintains proteostasis and membrane homeostasis through the activation of the ER stress sensor proteins"
VERIFIED (Attempt 2)
Source: ID: 42410967
"Reduced TFEB mRNA expression correlated with poor overall-survival and disease-free-survival across multiple cancer patients, with a particularly strong association in pancreatic cancer patients."
VERIFIED (Attempt 2)
Source: ID: 42410910
"In this study, we observed that sodium iodate (NaIO3), an oxidative stress inducer, triggered lysosomal dysfunction via lysosomal membrane permeabilization (LMP), thereby impairing autophagic flux in RPE cells and exacerbating retinal degeneration."
VERIFIED (Attempt 2)
Source: ID: 42410873
"Since drugs must pass through lipid bilayers to reach their targets, understanding the surface properties of biological membranes is crucial."
VERIFIED (Attempt 2)
Source: ID: 42412383
"Epigenetic mechanisms, including DNA methylation, histone modification, and microRNA-mediated regulation, may link chronological aging and cumulative environmental exposures to altered inflammatory signaling, extracellular matrix turnover, oxidative stress responses, and tissue repair."
VERIFIED (Attempt 2)
Source: ID: 42411514
"EA was associated with reduced markers of excessive autophagy and reduced ferroptosis markers in neurons."
VERIFIED (Attempt 2)
Source: ID: 42411331
"Berberine modulates ferroptosis via iron homeostasis, lipid peroxidation, the System Xc-/GSH/GPX4 anti-oxidant system, and mitochondrial function, involving NRF2, p53, AMPK, PI3K/Akt, and MAPK pathways."
VERIFIED (Attempt 2)
Source: ID: 42410294
"MAGMA identified enrichment for oxidative phosphorylation/mitochondrial electron transport chain, Notch, cell-cycle, DNA damage response-p53/TP53, and immune pathways."
VERIFIED (Attempt 2)
Source: ID: 42410284
"Cur protected against DA neuronal loss by modulating the interplay between cuproptosis and autophagy via the suppression of the AKT/mTOR/P70S6K."
VERIFIED (Attempt 2)
Source: ID: 42410256
"Additionally, SenFlag incorporates lysosomal features, including increased expression of V-ATPase subunits and cathepsins."
VERIFIED (Attempt 2)
Source: ID: 42411498
"Disruption of these systems promotes ADM formation and pancreatic intraepithelial neoplasia, whereas antioxidant treatment inhibits progression."
VERIFIED (Attempt 2)
Source: ID: 42411668
"As positive regulators, ncRNAs promote cell survival, regulate the cell cycle, enhance differentiation, selfrenewal, delay senescence, and modulate autophagy to maintain stem cell function."
VERIFIED (Attempt 2)
Source: ID: 42411477
"These enzymes share common features, including cofactor-dependent catalysis (NAD+/NADH or NADP+/NADPH), strategic positioning at metabolic nodes, and integration of compartmentalized metabolism between the cytosol and mitochondria."
VERIFIED (Attempt 2)
Source: ID: 42412246
"There is now abundant evidence that during aging and age-related diseases mitochondria are prone to release both mtDNA and dsRNA."
VERIFIED (Attempt 2)
Source: ID: 42411040
"THBS4 alleviated LPS-induced damage in NPCs through the activation of the PI3K/AKT pathway, exerting anti-apoptotic and anti-inflammatory effects; the specific upstream molecular mechanism of PI3K/AKT activation by THBS4 requires further investigation."
VERIFIED (Attempt 2)
Source: ID: 42411410
"NCDN associates with PRPF8-AAR2-EFTUD2 complex in the cytoplasm and is essential for the proper progression of this assembly intermediate toward mature U5 snRNP formation."
VERIFIED (Attempt 1)
Source: ID: 42412329
"mitophagy-a selective form of autophagy that clears damaged mitochondria-plays a crucial role in maintaining cellular homeostasis."
VERIFIED (Attempt 1)
Source: ID: 42148801
"In a recent publication, the authors identify Rpl12 and its homologs as receptors that promotes ribophagy from yeast to humans."
VERIFIED (Attempt 1)
Source: ID: 42383423
"Cells respond to this damage with an array of molecular countermeasures, ranging from membrane repair mechanisms to elimination of terminally damaged lysosomes by selective macroautophagy."
VERIFIED (Attempt 1)
Source: ID: 42410284
"Cur protected against DA neuronal loss by modulating the interplay between cuproptosis and autophagy via the suppression of the AKT/mTOR/P70S6K."
VERIFIED (Attempt 1)
Source: ID: 42410080
"CANA induced ATP deficiency and initiated autophagy, but concurrently impaired autophagosome-lysosome fusion."
VERIFIED (Attempt 1)
Source: ID: 42410967
"lysostilbene-4 induced rapid, irreversible lysosomal membrane permeabilization (LMP), initiating a lysosome mitochondria apoptotic cascade"
VERIFIED (Attempt 1)
Source: ID: 42396641
"TPLA protects against LPS‑induced systemic inflammation and hepatic‑pulmonary injury by modulating PINK1/Parkin‑associated mitophagy‑related signaling and macrophage polarization."
VERIFIED (Attempt 1)
Source: ID: 42397844
"EMCV sequentially activates non-redundant AKT3-dependent autophagic and apoptotic pathways to degrade TJ proteins"
VERIFIED (Attempt 1)
Source: ID: 42383423
"The limiting membrane of lysosomes is prone to damage that can have deleterious consequences for cellular homeostasis."
VERIFIED (Attempt 1)
Source: ID: 42411048
"LncRNA Mirt2 promoted chondrocyte proliferation and alleviated chondrocyte apoptosis, inflammation, and cartilage degeneration by activating miR-429/TBK1-mediated autophagy."
VERIFIED (Attempt 1)
Source: ID: 42397110
"Ursodeoxycholic Acid (UDCA) upregulated EGR1, which in turn promoted the expression of the autophagy-related protein LC3B and the lysosomal protein LAMP1, while reducing P62 accumulation."
VERIFIED (Attempt 1)
Source: ID: 42409845
"reduces epithelial secretion of IL-18 via mTOR-controlled secretory autophagy."
VERIFIED (Attempt 1)
Source: ID: 42385220
"DHJSD modulates autophagy-related markers via controlling the LC3-II/LC3-I ratio and BCL2, P62 expression."
VERIFIED (Attempt 1)
Source: ID: 42412302
"Resveratrol (RV), a natural polyphenolic compound, exhibits anti-aging properties through the regulation of autophagy and oxidative stress"
VERIFIED (Attempt 1)
Source: ID: 42410910
"sodium iodate (NaIO3), an oxidative stress inducer, triggered lysosomal dysfunction via lysosomal membrane permeabilization (LMP), thereby impairing autophagic flux"
VERIFIED (Attempt 2)
Source: ID: 42412329
"mitophagy-a selective form of autophagy that clears damaged mitochondria-plays a crucial role in maintaining cellular homeostasis."
VERIFIED (Attempt 2)
Source: ID: 42383423
"The limiting membrane of lysosomes is prone to damage that can have deleterious consequences for cellular homeostasis."
VERIFIED (Attempt 2)
Source: ID: 42383423
"Cells respond to this damage with an array of molecular countermeasures, ranging from membrane repair mechanisms to elimination of terminally damaged lysosomes by selective macroautophagy."
VERIFIED (Attempt 2)
Source: ID: 42148801
"In a recent publication, the authors identify Rpl12 and its homologs as receptors that promotes ribophagy from yeast to humans."
VERIFIED (Attempt 2)
Source: ID: 42412302
"Resveratrol (RV), a natural polyphenolic compound, exhibits anti-aging properties through the regulation of autophagy and oxidative stress"
VERIFIED (Attempt 2)
Source: ID: 42410080
"CANA induced ATP deficiency and initiated autophagy, but concurrently impaired autophagosome-lysosome fusion."
VERIFIED (Attempt 2)
Source: ID: 42410284
"Cur protected against DA neuronal loss by modulating the interplay between cuproptosis and autophagy via the suppression of the AKT/mTOR/P70S6K."
VERIFIED (Attempt 2)
Source: ID: 42410967
"lysostilbene-4 induced rapid, irreversible lysosomal membrane permeabilization (LMP), initiating a lysosome mitochondria apoptotic cascade"
VERIFIED (Attempt 2)
Source: ID: 42396641
"TPLA protects against LPS‑induced systemic inflammation and hepatic‑pulmonary injury by modulating PINK1/Parkin‑associated mitophagy‑related signaling and macrophage polarization."
VERIFIED (Attempt 2)
Source: ID: 42397844
"EMCV sequentially activates non-redundant AKT3-dependent autophagic and apoptotic pathways to degrade TJ proteins"
VERIFIED (Attempt 2)
Source: ID: 42411048
"LncRNA Mirt2 promoted chondrocyte proliferation and alleviated chondrocyte apoptosis, inflammation, and cartilage degeneration by activating miR-429/TBK1-mediated autophagy."
VERIFIED (Attempt 2)
Source: ID: 42397110
"Ursodeoxycholic Acid (UDCA) upregulated EGR1, which in turn promoted the expression of the autophagy-related protein LC3B and the lysosomal protein LAMP1, while reducing P62 accumulation."
VERIFIED (Attempt 2)
Source: ID: 42409845
"reduces epithelial secretion of IL-18 via mTOR-controlled secretory autophagy."
VERIFIED (Attempt 2)
Source: ID: 42385220
"DHJSD modulates autophagy-related markers via controlling the LC3-II/LC3-I ratio and BCL2, P62 expression."
VERIFIED (Attempt 2)
Source: ID: 42410910
"sodium iodate (NaIO3), an oxidative stress inducer, triggered lysosomal dysfunction via lysosomal membrane permeabilization (LMP), thereby impairing autophagic flux"
VERIFIED (Attempt 2)
Source: ID: 42352379
"reduced the expression of autophagy-related genes."
VERIFIED (Attempt 2)
Source: ID: 42353057
"suppression of autophagy can be detected in the blood of individuals with COPD"
VERIFIED (Attempt 2)
Source: ID: 42392747
"C7 primarily induced cell death by activating the autophagy pathway"
VERIFIED (Attempt 2)
Source: ID: 42389518
"autophagy levels in the intestine were significantly increased in SB group with enhanced protein levels of ATG16L1 and LC3-II, and reduced level of p62/SQSTM1 protein."
VERIFIED (Attempt 2)
Source: ID: 42377685
"there was autophagosome accumulation in transmission electron microscope (TEM)."
MISMATCH PRUNED (Attempt 1)
Source: ID: 42412329
"Mitophagy-a selective form of autophagy that clears damaged mitochondria-plays a crucial role in maintaining cellular homeostasis."
Validator Flag: Strict Misquote Detected! The exact character sequence "Mitophagy-a selective form of autop..." was NOT found in the provided text. Do NOT truncate, paraphrase, or edit quotes.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42410284
"Curcumin protected against DA neuronal loss by modulating the interplay between cuproptosis and autophagy via the suppression of the AKT/mTOR/P70S6K."
Validator Flag: Strict Misquote Detected! The exact character sequence "Curcumin protected against DA neuro..." was NOT found in the provided text. Do NOT truncate, paraphrase, or edit quotes.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42410080
"Mechanistically, CANA induced ATP deficiency and initiated autophagy, but concurrently impaired autophagosome-lysosome fusion. This dual effect led to autophagic flux blockade."
Validator Flag: Strict Misquote Detected! The exact character sequence "Mechanistically, CANA induced ATP d..." was NOT found in the provided text. Do NOT truncate, paraphrase, or edit quotes.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42409247
"Overexpression of CDKN1A in HD-MSNs alleviated HD pathologies... while improving neuronal survival and autophagy-associated activity."
Validator Flag: Ellipses (...) are strictly forbidden. You must quote continuous text exactly character-for-character.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42409090
"3-MCPD triggered Golgi stress, leading to Mitf-dependent transcriptional upregulation of Atg9. This, in turn, activated autophagy, which selectively degraded E-cadherin."
Validator Flag: Strict Misquote Detected! The exact character sequence "3-MCPD triggered Golgi stress, lead..." was NOT found in the provided text. Do NOT truncate, paraphrase, or edit quotes.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42407178
"Macrophage autophagy... plays a critical role in dynamically balancing immune responses by selectively eliminating damaged organelles, pathogens, and protein aggregates."
Validator Flag: Ellipses (...) are strictly forbidden. You must quote continuous text exactly character-for-character.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42404975
"CCL2 suppression enhanced neuronal autophagy, as evidenced by increased Beclin1 and LC3-II expression and modulation of autophagy flux markers (p62 and LAMP2)."
Validator Flag: Strict Misquote Detected! The exact character sequence "CCL2 suppression enhanced neuronal ..." was NOT found in the provided text. Do NOT truncate, paraphrase, or edit quotes.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42402931
"Mir452 significantly promotes protective autophagy in septic AKI by suppressing the APAF1-CASP9 axis."
Validator Flag: Strict Misquote Detected! The exact character sequence "Mir452 significantly promotes prote..." was NOT found in the provided text. Do NOT truncate, paraphrase, or edit quotes.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42402699
"CAMKV interacts with both RABV P and SQSTM1, promoting SQSTM1-mediated selective autophagic clearance of P."
Validator Flag: Strict Misquote Detected! The exact character sequence "CAMKV interacts with both RABV P an..." was NOT found in the provided text. Do NOT truncate, paraphrase, or edit quotes.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42410672
"At nanomolar concentrations, lysostilbene-4 induced rapid, irreversible lysosomal membrane permeabilization (LMP), initiating a lysosome mitochondria apoptotic cascade via CTSB (cathepsin B) release, BID cleavage, BAX activation, and caspase-mediated apoptosis."
Validator Flag: Quote was found in context but NOT in the specific abstract mapped to ID '42410672'.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42411471
"Recent studies have demonstrated that treatment with genistein ... which acts by stimulating autophagy through a FOXO3-dependent process, can ameliorate behavioral abnormalities."
Validator Flag: Ellipses (...) are strictly forbidden. You must quote continuous text exactly character-for-character.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42410080
"Mechanistically, CANA induced ATP deficiency and initiated autophagy, but concurrently impaired autophagosome-lysosome fusion. This dual effect led to autophagic flux blockade."
Validator Flag: Strict Misquote Detected! The exact character sequence "Mechanistically, CANA induced ATP d..." was NOT found in the provided text. Do NOT truncate, paraphrase, or edit quotes.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42411048
"LncRNA Mirt2 acted as a competing endogenous RNA (ceRNA) by sponging miR-429 in IL-1β-induced chondrocytes. Additionally, miR-429 directly targets TBK1."
Validator Flag: Strict Misquote Detected! The exact character sequence "LncRNA Mirt2 acted as a competing e..." was NOT found in the provided text. Do NOT truncate, paraphrase, or edit quotes.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42390723
"activation of the Nrf2 signaling pathway... promoting the initiation and execution of mitophagy to eliminate dysfunctional and damaged mitochondria"
Validator Flag: Ellipses (...) are strictly forbidden. You must quote continuous text exactly character-for-character.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42411514
"EA is associated with reduced markers of excessive autophagy and reduced ferroptosis markers in neurons."
Validator Flag: Strict Misquote Detected! The exact character sequence "EA is associated with reduced marke..." was NOT found in the provided text. Do NOT truncate, paraphrase, or edit quotes.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42412296
"The molecular pathogenesis of PD involves Oxidative stress... insufficient autophagy-lysosomal clearance, and synaptic degeneration"
Validator Flag: Ellipses (...) are strictly forbidden. You must quote continuous text exactly character-for-character.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42411996
"V-12c degraded both ERα and HDAC6 via the proteasome pathway... disrupted the autophagy-lysosome function"
Validator Flag: Ellipses (...) are strictly forbidden. You must quote continuous text exactly character-for-character.
MISMATCH PRUNED (Attempt 1)
Source: ID: 42410370
"Single-cell and spatial analyses revealed that the biologically significant FZD5 signal originated predominantly within tumor-associated macrophages (TAMs)... associated with mitophagy-related programs"
Validator Flag: Ellipses (...) are strictly forbidden. You must quote continuous text exactly character-for-character.
Mapped Reference Directory (APA)
- [1] ID: 42412300 - Liu D, Ma Y, Huang H, Cheng X, Ma Q et al. (2026). Dehydrocostus Lactone Activates Nrf2 Signaling Pathway to Attenuate Oxidative Stress, Inflammation, and Excessive Autophagy in a Mouse Model of Chronic Obstructive Pulmonary Disease.. Inflammation. ID: 42412300.
- [2] ID: 42409845 - Diab N, Yong CH, Stange EL, Birk MS, Schmitz MA et al. (2026). Salmonella SopB suppresses post-transcriptionally regulated cytokine release to reduce early tissue inflammation and delay disease progression.. Nature communications. ID: 42409845.
- [3] ID: 42409251 - Liu X, Yu S, Kang J, Kim JW (2026). Loss of ETV4 triggers PPM1E downregulation and AMPK-ULK1-dependent autophagy to promote intrinsic trametinib resistance in breast cancer.. Pharmacological research. ID: 42409251.
- [4] ID: 42409092 - Salem IS, Sadik NAH, Maurice NW, Abdel Rahman AAS, Mabrouk SS et al. (2026). L-Theanine attenuates isoprenaline-induced heart failure in rats through modulation of the JNK/c-Jun/Beclin 1/Bcl2 pathway.. Chemico-biological interactions. ID: 42409092.
- [5] ID: 42406081 - Ji L, Li K, Feng J, Yin JH, He YT et al. (2026). UBE2L6 promotes invasion and metastasis of triple-negative breast cancer by enhancing autophagy through STK38 ISGylation.. Cellular and molecular life sciences : CMLS. ID: 42406081.
- [6] ID: 42405585 - Zhang Z, Yu H, Hao L (2026). Targeted Inhibition of RPA3 Impairs Breast Cancer Progression Through Suppressing TGF-β Signaling Pathway-Mediated Autophagy.. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. ID: 42405585.
- [7] ID: 42405496 - Dutta A, Gurusubramanian G, Roy VK (2026). Zingerone ameliorates ovarian impairment by regulating steroidogenesis and apoptosis in letrozole-treated mouse.. Endocrinology. ID: 42405496.
- [8] ID: 42403159 - Alaçamlı G, Can N, Yakar K, Kaşıkcı M, Karalezli A (2026). Autophagy-related pathway dysregulation and apoptosis suppression in pterygium: Role of key biomarkers Beclin-1, ATG5, LC3, p62, and Bcl-2 in pathogenesis and potential nonsurgical targets.. The Journal of international medical research. ID: 42403159.
- [9] ID: 42402646 - Altabbal S, Hussein S, Iratni R, Al Dhaheri Y (2026). Acridocarpus orientalis ethanolic extract induces senescence and abortive autophagy in human breast cancer cells.. Scientific reports. ID: 42402646.
- [10] ID: 42401664 - Gebala P, Janowska J, Sypecka J (2026). Evaluation of autophagy in neonatal rat glial cells in an in vitro model of hypoxic-ischemic injury.. Scientific reports. ID: 42401664.
- [11] ID: 42401166 - Lu B, Liao N, Wang Y, Yang Z, Liu Y et al. (2026). Aloin suppresses cancer cell growth via autophagy-promoted NEDD8 de-NEDDylation to target GPX4 for ferroptosis induction.. Bioorganic chemistry. ID: 42401166.
- [12] ID: 42402699 - Wang J, Huo H, Tao Y, Wang J, Wang X et al. (2026). CAMKV is an ISG and facilitates the degradation of rabies virus phosphoprotein via SQSTM1-mediated selective autophagy.. Autophagy. ID: 42402699.
- [13] ID: 42401010 - Guo X, Cheng H, Wang D, Wang Z, Hu Q et al. (2026). COP9 signalosome 8 mediated autophagy drives proliferation, invasion, and metastasis in pancreatic ductal adenocarcinoma.. Biochemical and biophysical research communications. ID: 42401010.
- [14] ID: 42409090 - Liu Z, Song Y, Xie B, Xia P, Yang J et al. (2026). 3-monochloro-1,2-propanediol disrupts ovarian germline stem cell maintenance via Golgi stress-induced autophagy in Drosophila.. Chemico-biological interactions. ID: 42409090.
- [15] ID: 42410967 - Chauhan N, Guha Majumdar A, Bhutia SK, Dey P, Subramanian M et al. (2026). Targeting lysosomes with a novel chloroquine derivative induces irreversible lysosomal damage and disrupts autophagosome and lysosome assembly in cancer.. Autophagy. ID: 42410967.
- [16] ID: 42412415 - Glover E, Wiseman B, Dugdale C, Humphery C, Sueiro Ballesteros L et al. (2026). Intercellular mitochondrial transfer and trans-mitophagy in response to protein import dysfunction.. The Journal of cell biology. ID: 42412415.
- [17] ID: 42409767 - Labani-Motlagh A, Li Y, Gao DS, Lee E, Chen V et al. (2026). mTORC1 suppression by Trp53 mutation drives resistance to immune checkpoint blockade.. Cell death & disease. ID: 42409767.
- [18] ID: 42404975 - Jia Z, Zheng Y, Jin M, Qin H, Wang Y (2026). CCL2 regulates autophagy through the TNF signaling pathway to promote traumatic brain injury progression.. 3 Biotech. ID: 42404975.
- [19] ID: 42409247 - Lee SW, Upshaw TJ, Bailey DJ, Lee SA, Kim J et al. (2026). CDKN1A protects medium spiny neurons from Huntington's disease pathology.. Neurobiology of disease. ID: 42409247.
- [20] ID: 42402931 - Liu Z, Fu Y, Wu W, Cai J, Dong Z (2026). Mir452 protects against septic acute kidney injury by targeting Apaf1 to relieve CASP9-mediated suppression of autophagy.. Autophagy. ID: 42402931.
- [21] ID: 42412329 - Liu Z, Zhang S, Zeng T (2026). Mitophagy in Metabolic Dysfunction-Associated Fatty Liver Disease: Mechanisms, Regulatory Networks, and Therapeutic Perspectives.. Inflammation. ID: 42412329.
- [22] ID: 42412302 - Zhang C, Ma Y, Shi Z, He X, Hu J et al. (2026). Study on the Effects and Mechanisms of Resveratrol in Improving Cognitive Impairment in Aβ1-42-Induced Alzheimer's Disease Model Mice.. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. ID: 42412302.
- [23] ID: 42411996 - Xin L, Liu J, Guo X, Li S, Guo Z et al. (2026). Discovery of Effective Dual PROTAC Degraders with Synergistic Antitumor Activity for Overcoming Tamoxifen-Resistant Breast Cancer.. Journal of medicinal chemistry. ID: 42411996.
- [24] ID: 42411475 - Fujise K, Kimura H, Tsuji T, Kamikawa Y, Imaizumi K et al. (2026). Broad Roles of Endoplasmic Reticulum Stress Sensors Activated by Diverse Pathophysiological Stimuli.. Frontiers in bioscience (Landmark edition). ID: 42411475.
- [25] ID: 42410910 - Ji Y, Sun Y, Huang X, Liang J, Fang D et al. (2026). Targeting the SNAI1-LAMP3 axis to restore lysosomal function and alleviate autophagic flux impairment to delay retinal degeneration.. Autophagy. ID: 42410910.
- [26] ID: 42410873 - Zaborowska-Mazurkiewicz M (2026). Comparative Biophysical Analysis of Healthy and Inflamed Intestinal Membrane Models Using Langmuir Monolayers.. The journal of physical chemistry. B. ID: 42410873.
- [27] ID: 42412383 - Uehara O, Umeda K, Morikawa T, Yoshida K, Abiko Y (2026). Epigenetic mechanisms of gingival aging: biological basis, periodontal implications, and therapeutic perspectives.. Odontology. ID: 42412383.
- [28] ID: 42411514 - Li S, Gao L, Mu L, Lu J, Chen H et al. (2026). Electroacupuncture Alleviates Focal Cerebral Ischemia-Reperfusion Injury and Is Associated With Modulation of Autophagy-Ferroptosis Involving the STAT3/HIF-1α Signalling Pathway.. Clinical and experimental pharmacology & physiology. ID: 42411514.
- [29] ID: 42411331 - Cheng T, Xi H, Hao W, Yang Y, Qian N et al. (2026). Targeting Ferroptosis: Effects and Mechanisms of Action of Berberine.. The American journal of Chinese medicine. ID: 42411331.
- [30] ID: 42410294 - Zheng J, Wang C, Xu J, Lu R, Gao H (2026). Shared genetic liability between opioid analgesic use and lung cancer subtypes revealed by genome wide correlation and structural modeling.. Discover oncology. ID: 42410294.
- [31] ID: 42410284 - Ren F, Sun Y, Wang M, Zheng K, Zuo C et al. (2026). Curcumin Attenuates Cuproptosis via Activating Autophagy Through Inhibition of the AKT/mTOR/P70S6K-Signaling Pathway in Parkinson's Disease Models.. Molecular neurobiology. ID: 42410284.
- [32] ID: 42410256 - Altulea A, Mackedenski S, Nehme J, Demaria M (2026). SenFlag gene signature identifies senescent cells in mouse and human tissues through a conserved core transcriptional program.. The EMBO journal. ID: 42410256.
- [33] ID: 42411498 - Qi DY, Jin WL (2026). Navigating the Redox Precipice: Metabolic Gatekeeping as a Therapeutic Window in Pancreatic Precancer.. Frontiers in bioscience (Landmark edition). ID: 42411498.
- [34] ID: 42411668 - Alfaifi M (2026). The Dual Role of Non-Coding RNAs in Regulating HSC and MSC Fate: Modulating Survival, Death, and Intercellular Communication.. Clinical laboratory. ID: 42411668.
- [35] ID: 42411477 - Papaneophytou C, Petrou C (2026). Six Dehydrogenase Gatekeepers of Carbohydrate Metabolism: Metabolic Integration in Health and Disease.. Frontiers in bioscience (Landmark edition). ID: 42411477.
- [36] ID: 42412246 - Salminen A, Kaarniranta K, Kauppinen A (2026). Endosymbiotic theory of aging revisited: Age-related leakage of mitochondrial dsDNA/RNA stimulates cytosolic nucleic acid sensors which remodel the immune network and promote the aging process.. Biogerontology. ID: 42412246.
- [37] ID: 42411040 - Shu Y, Sun C, Chen S (2026). Thrombospondin-4 Regulates Lipopolysaccharide-Induced Apoptosis and Inflammation in Nucleus Pulposus Cells via the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway.. Immunity, inflammation and disease. ID: 42411040.
- [38] ID: 42411410 - Ren T, Huang W, Wei G, Zhao H, Zhang Y et al. (2026). Neurochondrin promotes U5 snRNP maturation by regulating AAR2 release from PRPF8.. Nucleic acids research. ID: 42411410.
- [39] ID: 42383423 - Meyer H, Kuma A, Nakamura S (2026). Disentangling the response to lysosomal damage.. Journal of cell science. ID: 42383423.
- [40] ID: 42148801 - Govind CK, Klionsky DJ (2026). Identification of a conserved receptor for degrading ribosomes through autophagy.. Autophagy. ID: 42148801.
- [41] ID: 42410080 - Wang Y, Zhang E, Ma R, Liu W, Wang Q et al. (2026). SGLT2 inhibition induces autophagic flux blockade and sensitizes pancreatic cancer to EGFR-targeted therapy.. Cellular oncology (Dordrecht, Netherlands). ID: 42410080.
- [42] ID: 42396641 - Wei J, Zhong W, Zhou G, Huang M, Huang X et al. (2026). Laggera alata attenuates LPS‑induced systemic inflammation and hepatic‑pulmonary injury by regulating PINK1/Parkin-associated mitophagy and macrophage polarization.. Molecular medicine reports. ID: 42396641.
- [43] ID: 42397844 - Dou X, Wang N, Yao S, Chen X, Li S et al. (2026). Encephalomyocarditis virus impairs the blood-brain barrier by degrading tight junction proteins via AKT3-dependent autophagic and apoptotic pathways.. Virulence. ID: 42397844.
- [44] ID: 42411048 - Zhang JX, Li Z, Yang P, Pu K, Zhou Q et al. (2026). LncRNA Mirt2 Attenuates Osteoarthritis Progression by Promoting miR-429/TBK1-Mediated Autophagy.. Immunity, inflammation and disease. ID: 42411048.
- [45] ID: 42397110 - Zhang Y, Han Q, Liu Y, Shen W, Cheng S et al. (2026). EGR1 Mediates Ursodeoxycholic Acid-Promoted Mitophagy to Prevent Postovulatory Aging of Porcine Oocytes.. Aging cell. ID: 42397110.
- [46] ID: 42385220 - Song C, Wu X, Chen C, Wang X, Liu F et al. (2026). Duhuo Jisheng Decoction Mitigates Intervertebral Disc Degeneration via Metabolic Reprogramming and TGF-β/Smad-Driven Autophagy-Fibrosis Network Modulation.. Biomedical chromatography : BMC. ID: 42385220.
- [47] ID: 42352379 - Ferrini F, Annibalini G, Battistelli M, Moosavi S, Riham O et al. (2026). Revitalizing Muscle Repair: Hyaluronan Preserves Mitochondrial Architecture and Promotes Myogenesis Under Pro-Inflammatory Conditions.. Biomolecules. ID: 42352379.
- [48] ID: 42353057 - Cooper JM, Chen S, Lester SE, Kim J, Gummow J et al. (2026). Autophagy Is Suppressed in Peripheral Blood Mononuclear Cells During Chronic Obstructive Pulmonary Disease.. International journal of molecular sciences. ID: 42353057.
- [49] ID: 42392747 - Shen SY, Zhang JW, Liu MH, Liu J, Tian WJ et al. (2026). [Mechanistic investigation of a natural compound against tumors via modulation of nuclear receptor RXRα-mediated autophagy pathway].. Zhongguo Zhong yao za zhi = Zhongguo zhongyao zazhi = China journal of Chinese materia medica. ID: 42392747.
- [50] ID: 42389518 - Mao Q, Lin B, Zhang W, Zhang Y, Lei Y et al. (2026). Microbiota metabolite butyrate alleviates intestinal inflammation associated with enhanced autophagy-related signaling in DSS-induced colitis.. Frontiers in immunology. ID: 42389518.
- [51] ID: 42377685 - Abass SA, Abdelrafea R, Eldomany RA, Elsisy RA, Zakaria S (2026). Clomipramine potentiates sorafenib efficacy in experimental hepatocellular carcinoma by targeting lysosomal sequestration and modulating the cathepsin B/Bcl-2/Beclin-1 axis.. Molecular biology reports. ID: 42377685.
Abstract Repository (Raw Full-Texts) Show Database Collapse Database
REFERENCE [40] · ID: 42148801
ID: 42148801 Title: Identification of a conserved receptor for degrading ribosomes through autophagy. Abstract: Ribosomes consist of approximately 80 distinct ribosomal proteins and rRNA. The genes encoding these ribosomal components are among the most highly expressed in growing cells. Changes in ribosome composition, such as those induced by oxidative stress, may compromise ribosome function. Such ribosomes are subsequently targeted for degradation. Additionally, under stress, both protein synthesis and ribosome biogenesis are downregulated. Under starvation stress, excess ribosomes are degraded through a process called ribophagy, a selective form of macroautophagy/autophagy that utilizes the autophagy pathway. While receptors for several selective autophagy pathways are known, the evolutionarily conserved ribophagy receptor was not identified until recently. In a recent publication, the authors identify Rpl12 and its homologs as receptors that promotes ribophagy from yeast to humans. They also demonstrate that ribophagy enhances lifespan and facilitates the clearance of pathogenic bacteria.Abbreviations: AIM: Atg8-family interacting motif; ATG: autophagy related; LIR: LC3-interacting region; NUFIP1: nuclear FMR1 interacting protein 1.
REFERENCE [47] · ID: 42352379
ID: 42352379 Title: Revitalizing Muscle Repair: Hyaluronan Preserves Mitochondrial Architecture and Promotes Myogenesis Under Pro-Inflammatory Conditions. Abstract: Hyaluronic acid (HA), a major component of the glycome and a non-sulfated glycosaminoglycan, plays a crucial role in regulating stem cell behavior and function, thereby supporting skeletal muscle repair under inflammatory conditions. In this study, we investigated the effects of a mixture of HA fractions with different molecular weights (M-HA; 2-1000 kDa) on the repair capacity and myogenic potential of C2C12 murine myoblasts exposed to inflammatory stimuli. C2C12 cells were cultured, induced to differentiate, and treated with M-HA (1 mg/mL) under either physiological or inflammatory conditions (LPS, 10 µg/mL; IL-1β, 20 ng/mL). M-HA exhibited no cytotoxic effects, even at the highest concentration tested (1.0 mg/mL), and significantly enhanced scratch wound closure. Moreover, M-HA improved the myogenic index at day 5 of differentiation, promoted the expression of myogenic markers, preserved myosin heavy chain (MHC) levels under inflammatory stress, and reduced the expression of autophagy-related genes. Ultrastructural analyses revealed that untreated myotubes displayed swollen mitochondria, disrupted cristae architecture, and numerous autophagic vacuoles, whereas M-HA-treated cells exhibited well-preserved mitochondrial morphology, intact cristae organization, reduced cytoplasmic damage, and maintained myofibrillar structure. Taken together, the functional, molecular, and ultrastructural findings demonstrate that M-HA protects myoblasts from inflammation-induced cellular damage and supports their regenerative capacity. These results underscore the potential of glycomics-based strategies to enhance myogenic differentiation and promote skeletal muscle regeneration in inflammatory microenvironments.
REFERENCE [48] · ID: 42353057
ID: 42353057 Title: Autophagy Is Suppressed in Peripheral Blood Mononuclear Cells During Chronic Obstructive Pulmonary Disease. Abstract: Assessing autophagy may offer insights into the pathogenesis of chronic obstructive pulmonary disease (COPD). However, measuring the dynamic aspect of autophagy is challenging, and sample manipulation can cause signal fluctuations that deviate from physiological conditions. We applied an organotypic method to quantify autophagy in COPD, where it frequently demonstrates disease-related dysregulation. Blood from control and COPD participants was treated with or without chloroquine. Microtubule-associated protein 1 light chain 3B II (LC3B-II) abundance was quantified in peripheral blood mononuclear cells (PBMCs), and findings were validated by transmission electron microscopy. Our observations show that while basal LC3B-II abundance was similar between groups (p = 0.60), autophagic flux was significantly lower in the COPD cohort, suggesting disruption in the regulatory factors that direct autophagosome clearance (p = 0.004). This was supported by less frequent observations of autophagy-related vacuoles in the cytosol of COPD-derived PBMCs. Our findings indicate that the suppression of autophagy can be detected in the blood of individuals with COPD, which warrants further investigation into its contribution to extrapulmonary disease processes.
REFERENCE [51] · ID: 42377685
ID: 42377685 Title: Clomipramine potentiates sorafenib efficacy in experimental hepatocellular carcinoma by targeting lysosomal sequestration and modulating the cathepsin B/Bcl-2/Beclin-1 axis. Abstract: Hepatocellular carcinoma (HCC) is one of the most common causes of death around the world, and it is commonly diagnosed late, so systemic chemotherapy is commonly used. Lysosomal sequestration of chemotherapy drugs such as sorafenib [1] decreases the effective concentration of chemotherapy at the target sites and leads to chemoresistance, also chemoresistance may arise from autophagy activation. We aimed to find a new therapeutic regimen for HCC through enhancing the chemosensitivity of SB by combining it with the lysosomotropic drug clomipramine (CM). 48 healthy Wister albino male rats were included. Induction of experimental HCC was done by intraperitoneal injection of diethylnitrosamine (DENA) (200 mg/ kg), after that, phenobarbital sodium (0.05%) was added to drinking water for 18 weeks. After induction, SB (10 mg/kg) was taken orally for 21 days. CM (10 mg/kg) was also provided orally for 21 days. The combination group received SB and CM (10 mg/kg) for 21 days. Treatment by CM in combination with SB could decrease neoplastic features in HCC group. The hepatic expression of Bcl2 was decreased, and the release of cytosolic cathepsin B was increased in the combination group. Also, the hepatic concentration of Beclin-1 decreased in the combination group and there was autophagosome accumulation in transmission electron microscope (TEM). The results indicate that the concomitant use of CM and SB may be considered a possible new therapeutic option in managing HCC by targeting the cathepsin B/Bcl2/Beclin-1 pathway.
REFERENCE [39] · ID: 42383423
ID: 42383423 Title: Disentangling the response to lysosomal damage. Abstract: The limiting membrane of lysosomes is prone to damage that can have deleterious consequences for cellular homeostasis. Cells respond to this damage with an array of molecular countermeasures, ranging from membrane repair mechanisms to elimination of terminally damaged lysosomes by selective macroautophagy. The various elements of this response therefore need to be carefully assessed in the context of the specific pathological or experimental conditions being studied. Emerging evidence has revealed further complexity within the lysosomal damage response, such as processes that contribute to initial membrane resealing as well as lysosome regeneration required to restore the lysosomal system. These mechanisms involve unusual ubiquitylation, non-canonical ATG8 lipidation, or modifications that govern lysosome tubulation or microlysophagy pathways. Therefore, caution is advised when using previously established lysosome damage reporters that might confound interpretation of the underlying events and outcomes. This Opinion article seeks to shed light on the emerging regulatory mechanisms of lysosomal regeneration and evaluate the appropriateness of various reporters and assays for studying the lysosomal damage response.
REFERENCE [46] · ID: 42385220
ID: 42385220 Title: Duhuo Jisheng Decoction Mitigates Intervertebral Disc Degeneration via Metabolic Reprogramming and TGF-β/Smad-Driven Autophagy-Fibrosis Network Modulation. Abstract: Duhuo Jisheng Decoction (DHJSD) shows promise for treating intervertebral disc degeneration (IVDD), but its mechanisms concerning autophagy and fibrosis are unclear. Using network pharmacology, metabolomics, UHPLC-Q-TOF/MS, and functional studies (in vitro and in vivo), we systematically explored DHJSD's molecular mechanisms. DHJSD has 254 constituents; those may regulate inflammation, apoptosis, and metabolic processes. DHJSD attenuates ECM/fibrosis-related changes, lowers BMP2 expression, is associated with reduced TGF-β/Smad2/3 phosphorylation, and partially improves annulus fibrosus morphology. SB431542 attenuated IL-1β-induced TGF-β pathway activation and BMP2 expression, supporting the involvement of this pathway in DHJSD-related regulation of fibrosis markers. The levels of serum IL-1β and TNF-α significantly decreased in animal models. Through glycerophospholipid and sphingolipid metabolism, DHJSD reshapes lipid homeostasis and may be associated with reduced TGF-β overactivation by downregulating pro-fibrotic compounds and upregulating anti-inflammatory metabolites. DHJSD modulates autophagy-related markers via controlling the LC3-II/LC3-I ratio and BCL2, P62 expression. DHJSD may affect glycolysis-related and oxidative phosphorylation-related changes and may be associated with phosphatidylcholine/ethanolamine-related mitochondrial membrane changes. DHJSD treats IVDD via a "metabolic reprogramming-TGF-β-related regulation-autophagy/mitochondrial-related remodeling" network, suggesting a potential multi-target strategy and demonstrating the value of multi-omics in analyzing traditional medicine.
REFERENCE [50] · ID: 42389518
ID: 42389518 Title: Microbiota metabolite butyrate alleviates intestinal inflammation associated with enhanced autophagy-related signaling in DSS-induced colitis. Abstract: The incidence of inflammatory bowel disease (IBD) has been demonstrated to be increased over recent decades. Butyrate derived from the gut microbiota is known to be beneficial in alleviating inflammation, yet the underlying mechanisms remain undefined. Human and mice fecal samples were analyzed using gas chromatography-mass spectrometry and 16S rRNA gene sequencing. Male wild-type C57BL/6J mice aged 6-8 weeks old were administered dextran sodium sulfate (DSS) to induce experimental colitis models. Mice were treated with sodium butyrate (SB) through oral gavage. 3-methyladenine (3MA) was administered intraperitoneally to suppress autophagy in mice. Our results showed that the butyric acid level in the feces of IBD patients was significantly lower than those in healthy controls (HCs) (134.5 vs. 605.9, p = 0.002), concomitant with a deficiency in butyrate-producing probiotics, such as Faecalibacterium. We found that oral SB changed the composition of the intestinal microbes (higher abundance of Barnesiella), restored intestinal barrier function determined by enhanced tight junction protein expression (OCCLUDIN) in Western blotting and diminished the susceptibility of mice to DSS-induced colitis. Additionally, autophagy levels in the intestine were significantly increased in SB group with enhanced protein levels of ATG16L1 and LC3-II, and reduced level of p62/SQSTM1 protein. While the SB group showed changes consistent with enhanced autophagy-related signaling, 3MA-treated mice conversely displayed significantly attenuated autophagy activity. Meanwhile, the butyrate-mediated protection against colonic injury was considerably diminished in the 3MA-treated mice. Our findings provide multi-line evidence that SB coordinates gut microbiota and is associated with enhanced autophagy-related signaling to alleviate inflammation in DSS-induced colitis, integrating human fecal metabolomic and microbiome analyses with in vivo pharmacological and transcriptomic data.
REFERENCE [49] · ID: 42392747
ID: 42392747 Title: [Mechanistic investigation of a natural compound against tumors via modulation of nuclear receptor RXRα-mediated autophagy pathway]. Abstract: Cancer treatment urgently requires individualized and precise strategies, and the development of highly selective drugs targeting specific molecular targets has become the core direction of current research. This study focused on the antitumor activity of the flavonoid compound cudratricusxanthone E(CAS 740810-46-2, C7), finding that it can significantly inhibit the proliferation of human cervical cancer HeLa cells in a time-dependent manner. Through the intervention of different cell death inhibitors, this study preliminarily revealed the potential pathway by which C7 induced cell death. The experiments found that the autophagy inhibitor chloroquine effectively blocked C7-mediated cell death, whereas the apoptosis inhibitor z-Val-Ala-Asp(OMe)-fluoromethylketone(Z-VAD-FMK) and the necroptosis inhibitor necrostatin-1(Nec-1) showed no significant effect. This suggested that C7 primarily induced cell death by activating the autophagy pathway, rather than through apoptosis or necroptosis, providing a key clue for understanding the compound's mechanism of action. To further elucidate its molecular mechanism, the study combined network pharmacology predictions with dual-luciferase reporter gene assays, identifying for the first time that the retinoid X receptor α(RXRα) was the target of C7. RXRα is a key regulatory factor in the nuclear receptor family, playing multiple roles in cell proliferation, differentiation, and metabolic regulation. In recent years, it has also been found to have regulatory significance in certain tumor processes. Subsequent experiments confirmed that C7 specifically bound to RXRα, triggering the phosphorylation of downstream adenosine monophosphate-activated protein kinase(AMPK). The activation of AMPK, as a central hub in cellular energy homeostasis and autophagy initiation, significantly promoted autophagic flux. Therefore, C7 drove autophagic cell death in HeLa cells by activating the RXRα/AMPK signaling axis, thereby exerting its antitumor effects. In summary, this study systematically elucidates the novel mechanism by which C7 induces tumor cell death, revealing the complete signaling pathway from the compound targeting RXRα to AMPK activation and ultimately leading to autophagic cell death.
REFERENCE [42] · ID: 42396641
ID: 42396641 Title: Laggera alata attenuates LPS‑induced systemic inflammation and hepatic‑pulmonary injury by regulating PINK1/Parkin-associated mitophagy and macrophage polarization. Abstract: Laggera alata is a traditional medicinal herb used for inflammatory and infectious diseases, but its mechanisms against endotoxin‑induced systemic inflammation remain unclear. The present study investigated the protective effects of total phenolics from Laggera alata (TPLA) on lipopolysaccharide (LPS)‑induced inflammatory injury and explored the involvement of PTEN‑induced putative kinase 1 (PINK1)/Parkin‑associated mitophagy and macrophage polarization. LPS‑induced inflammatory models were established in RAW264.7 macrophages and C57BL/6 mice. Cell viability, apoptosis, mitochondrial membrane potential (MMP), cytokine production, macrophage polarization and mitophagy‑related protein expression were evaluated. Mdivi‑1 was used to assess the involvement of mitophagy‑related signaling. In vivo, core body temperature, serum cytokines, and lung and liver histopathology were examined. TPLA improved the viability of LPS‑stimulated macrophages, reduced apoptosis, restored MMP, decreased p62 expression, and increased PINK1, Parkin and the LC3‑II/LC3‑I ratio. TPLA also suppressed M1‑associated indicators, including inducible nitric oxide synthase, IL‑12 and CD80/CD86, while enhancing M2‑associated indicators, including arginase 1, IL‑10 and CD206/CD163. In addition, TPLA reduced IL‑1β, IL‑6 and TNF‑α release. Mdivi‑1 partially reversed the effects of high‑dose TPLA on mitophagy‑related protein expression and macrophage polarization. In LPS‑challenged mice, TPLA alleviated hypothermia, reduced systemic cytokine levels, and attenuated hepatic and pulmonary injury. These findings suggest that TPLA protects against LPS‑induced systemic inflammation and hepatic‑pulmonary injury by modulating PINK1/Parkin‑associated mitophagy‑related signaling and macrophage polarization.
REFERENCE [45] · ID: 42397110
ID: 42397110 Title: EGR1 Mediates Ursodeoxycholic Acid-Promoted Mitophagy to Prevent Postovulatory Aging of Porcine Oocytes. Abstract: Postovulatory oocyte aging (POA) is a key factor contributing to the decline in female fertility and the success rate of assisted reproductive technology. Currently, most studies on POA have focused on downstream phenotypes such as mitochondrial dysfunction and oxidative stress, while little is known about its key upstream regulatory factors. Here, we show that the downregulation of transcription factor Early Growth Response 1 (EGR1) is a key upstream event driving porcine oocyte aging. Microtranscriptome sequencing combined with experimental validation verified a notable reduction in EGR1 protein abundance in aged oocytes. We found that Ursodeoxycholic Acid (UDCA) upregulated EGR1, which in turn promoted the expression of the autophagy-related protein LC3B and the lysosomal protein LAMP1, while reducing P62 accumulation. Furthermore, UDCA enhanced the expression of mitophagy core proteins PINK1, VDAC1 and promoted mitochondrial-lysosomal colocalization, thereby improving mitophagy and restoring the quality of aged oocytes. Crucially, treatment with the EGR1 inhibitor plicamycin completely blocked UDCA's ability to enhance the developmental potential of aged oocytes, confirming that EGR1-mediated mitophagy was the core pathway underlying UDCA's effects. Collectively, this study innovatively identified EGR1 as a key bridge linking oocyte aging and decreased mitophagy, and clarified the novel mechanism by which UDCA exerts its protective effects through the "UDCA-EGR1-mitophagy" axis. Our findings advanced the research on oocyte aging from phenotypic observation to the upstream transcriptional regulation level, providing a novel theoretical target and experimental basis for fundamentally intervening in reproductive aging.
REFERENCE [43] · ID: 42397844
ID: 42397844 Title: Encephalomyocarditis virus impairs the blood-brain barrier by degrading tight junction proteins via AKT3-dependent autophagic and apoptotic pathways. Abstract: Encephalomyocarditis virus (EMCV) infection causes viral encephalitis; however, the mechanisms underlying blood-brain barrier (BBB) disruption remain poorly understood. Here, we demonstrate that EMCV actively replicates in mouse brain tissue, induces robust neuroinflammation characterized by elevated proinflammatory cytokines and chemokines, and markedly increases BBB permeability as evidenced by Evans blue and sodium fluorescein extravasation. Importantly, tight junction (TJ) proteins ZO-1 and Occludin are selectively degraded at the post-transcriptional level, whereas Claudin-5 expression remains stable. Consistently, in vitro BBB models confirmed EMCV traversal, reduced transendothelial electrical resistance, and TJ disruption. Mechanistically, EMCV induces biphasic PI3K/AKT modulation and specifically downregulates AKT3. Notably, AKT3 knockdown exacerbates both autophagy and apoptosis, thereby accelerating ZO-1 and Occludin degradation while promoting viral replication. Furthermore, pharmacological inhibition of autophagy (chloroquine) or apoptosis (Z-VAD-FMK) effectively rescues TJ proteins and reduces viral load. Interestingly, the Caspase-8 inhibitor Z-IETD-FMK provides the most robust protection, implicating the extrinsic apoptotic pathway as the dominant route. Collectively, EMCV sequentially activates non-redundant AKT3-dependent autophagic and apoptotic pathways to degrade TJ proteins, ultimately enabling viral traversal across the compromised BBB and offering therapeutic targets for viral encephalitis.
REFERENCE [13] · ID: 42401010
ID: 42401010 Title: COP9 signalosome 8 mediated autophagy drives proliferation, invasion, and metastasis in pancreatic ductal adenocarcinoma. Abstract: The present study aimed to explore the role of COP9 signalosome 8 (COPS8) as a novel molecule in pancreatic ductal adenocarcinoma (PDAC). A total of 9 genes were first identified by intersecting the differential genes of the GSE15471 and GSE62165 datasets with 248 Neddylation genes from the Reactome Pathway Database. The association between disease-free survival and the 9 genes in patients with PDAC was analyzed. Analysis using The Cancer Genome Atlas, Gene Expression Omnibus and Gene Expression Profiling Interactive Analysis databases revealed that COPS8 was highly expressed in patients with PDAC, and PDAC tissues exhibited significantly higher COPS8 expression levels compared to those found in paracancerous tissues. Finally, the above results were verified by cellular experiments, reverse transcription-quantitative PCR, Western blotting and immunohistochemistry. The mRNA expression levels of COPS8 were significantly elevated in the pancreatic cancer cell lines PANC-1and MIA PaCa-2 compared to those in HPNE normal pancreatic cells, and the protein expression levels of COPS8 were also significantly elevated in the pancreatic cancer cells PANC-1 and MIA PaCa-2. COPS8 protein was significantly increased in cancer tissues of patients with pancreatic cancer compared to paracancerous tissues. The proliferative, migratory and invasive abilities of PANC-1 and MIA PaCa-2 cells were significantly reduced after knockdown of COPS8. The results showed that knockdown of COPS8 in PANC-1 and MIA PaCa-2 cells decreased Microtubule-associated proteins 1A/1B light chain 3B (LC3Ⅱ/LC3Ⅰ), while Sequestosome 1 (P62/SQSTM1) expression was elevated. COPS8 may promote the proliferation, invasion, and metastasis of pancreatic cancer cells by regulating autophagy.
REFERENCE [11] · ID: 42401166
ID: 42401166 Title: Aloin suppresses cancer cell growth via autophagy-promoted NEDD8 de-NEDDylation to target GPX4 for ferroptosis induction. Abstract: Aloin (ALO), an anthraquinone derived from Aloe vera, exhibits antitumor activity; however, its precise mechanisms of action remain unclear. In this study, in silico molecular docking analysis first revealed that Aloin (ALO) bound effectively to ferroptosis-related proteins (SLC7A11, GPX4, ACSL4, and TFR1). Subsequently, in vitro ALO treatment triggered ferroptosis hallmarks in human cervical cancer cell line Hela and mouse colon cancer cell line MC38 at both transcriptional and protein levels-downregulating SLC7A11/GPX4, upregulating ACSL4/TFR1, with Fe2+/ROS accumulation, GSH depletion, and ferroptosis-specific mitochondrial cristae loss. Furtherly, ALO inhibited cancer cell proliferation, migration, and invasion, effects that were reversed by the ferroptosis inhibitor Ferrostatin-1. Concurrently, ALO induced autophagy, as evidenced by increased levels of LC3, LaminB1, and ULK1, decreased levels of P62, and TEM-visualized autophagosomes. Notably, the autophagy inhibitor chloroquine reversed ALO-induced ferroptosis, NEDD8 downregulation, and NEDP1 upregulation, linking ALO-induced autophagy to NEDD8 de-NEDDylation. Genetic and pharmacological perturbation of the NEDD8 pathway confirmed this: NEDD8 inhibition enhanced, while NEDP1 knockdown attenuated, ALO-induced ferroptosis. Co-immunoprecipitation and laser-scanning confocal microscope confirmed a direct NEDD8-GPX4 interaction diminished by ALO, positioning GPX4 as a key effector. In in vivo study, ALO effectively inhibited cancer cell growth in a murine colon carcinoma MC38 xenograft models, while exhibiting no obvious toxicity or side effects in mice. Moreover, ALO exhibited the same regulatory effects and trends on ferroptosis-related proteins in vivo as those in vitro. In summary, this study reveals a novel mechanism that ALO-induced autophagy promotes NEDD8 de-NEDDylation, driving ferroptosis via the NEDD8-GPX4 axis to suppress cancer cell growth.
REFERENCE [10] · ID: 42401664
ID: 42401664 Title: Evaluation of autophagy in neonatal rat glial cells in an in vitro model of hypoxic-ischemic injury. Abstract: The autophagy process is crucial for cell functioning, yet it is still understudied in glial cells during neurodevelopment. To address this, cultures of the main glial cell types in the central nervous system (CNS), including astrocytes, microglia, oligodendrocyte progenitors, and differentiating oligodendrocytes, were created to examine the impact of an in vitro hypoxia-ischemia (HI) model on autophagy. The HI insult was mimicked by applying temporal oxygen-glucose deprivation (OGD). Since neonatal hypoxic-ischemic insults primarily affect the brain's white matter, the study predominantly focused on oligodendrocytes at different stages of maturation: progenitor cells versus cells that express myelin components (e.g. MBP). The results show that the different glial fractions exhibit varying sensitivity to the applied conditions. Maturing oligodendrocytes were found to be more sensitive to OGD conditions than the progenitor fraction. The OGD procedure was proven to impact the expression of autophagy markers, indicating the activity of this process in response to injury. Western blot analysis of oligodendrocyte progenitor cells (OPCs) showed that the autophagy substrate marker p62 increased after six hours, which may suggest transient inhibition and subsequent activation of autophagy. To verify the involvement of autophagy in the differentiation of neonatal oligodendrocytes, the process was modulated using chloroquine (CQ) treatment. CQ is recognised as an inhibitor of autophagic flux because it disrupts lysosomal acidity and prevents the breakdown of autophagosomes. CQ treatment resulted in the accumulation of autophagosomes. The results suggest that abnormalities in the functioning of glial cells, particularly oligodendrocytes, in response to hypoxic-ischaemic (HI)-like conditions might be associated with altered autophagic flux in response to cellular stress. Transient alterations in autophagy were observed within 24 h of limiting oxygen and glucose supply, and these alterations may contribute to subsequent disorders in oligodendrocyte differentiation. This is recognised as one of the major issues in the pathogenesis of neonatal hypoxia-induced damage. Therefore, modulation of autophagy could be a promising therapeutic approach to prevent these adverse changes.
REFERENCE [9] · ID: 42402646
ID: 42402646 Title: Acridocarpus orientalis ethanolic extract induces senescence and abortive autophagy in human breast cancer cells. Abstract: Breast cancer is the most frequently diagnosed cancer in women worldwide. Triple Negative Breast Cancer (TNBC), which lacks the expression of the hormonal Estrogen Receptor (ER) and Progesterone Receptor (PR), amplification of Human Epidermal Growth Factor Receptor 2 (HER2), is not responsive to the hormonal therapy. Currently, available chemotherapy and radiotherapy cause severe side effects; therefore, there is an urgent need for new therapeutic choices for TNBC. Acridocarpus orientalis is used in folk medicine to treat several health conditions. Here, evaluated the anti-cancer activity of Acriodocarpus orientalis Ethanolic Extract (AOEE) against two TNBC (MDA-MB-231 and Hs578T) and one luminal A (MCF-7) cell lines, and investigated the molecular mechanisms underlying its anticancer activity. The results revealed that AOEE inhibited cell proliferation of the three cell lines in a concentration- and time-dependent manner. The anti-proliferative effect of AOEE was found to be concomitant with the induction of cell cycle arrest at the G1/S phase. These changes were associated with upregulation of p21WAF1 and p27 Kip1, downregulation of PCNA, Cyclin D1, phospho-Rb. Moreover, AOEE induces abortive autophagy through upregulation of autophagy related proteins LC3-II, Beclin-1, and p62. Also, p16-dependent senescence was induced in AOEE treated MDA-MB-231 cells confirmed by senescence-associated β-galactosidase (SA-β-gal) expression in the treated cells. AOEE induced activation of ERK and p38 pathways, which might be involved in autophagy and senescence induction. Acridocarpus orientalis could be a potential source for novel chemotherapeutic agents against TNBC.
REFERENCE [12] · ID: 42402699
ID: 42402699 Title: CAMKV is an ISG and facilitates the degradation of rabies virus phosphoprotein via SQSTM1-mediated selective autophagy. Abstract: Once rabies virus (RABV) gains access to the central nervous system, infection almost inevitably results in fatal outcomes, and our incomplete understanding of viral pathogenesis remains a major barrier to effective therapeutic intervention. Here, we identify CAMKV as an interferon-stimulated gene (ISG) that drives the macroautophagic/autophagic degradation of RABV phosphoprotein (P), thereby potently suppressing viral replication in vitro. Notably, in vivo overexpression of CAMKV significantly delays disease progression in mice challenged with a street strain of RABV. Mechanistically, CAMKV interacts with both RABV P and SQSTM1, promoting SQSTM1-mediated selective autophagic clearance of P and thereby restricting RABV transcription and replication. Collectively, our findings establish CAMKV as a critical host antiviral effector that functions through selective autophagy, highlighting CAMKV as a promising molecular target for the development of novel therapeutics against lethal RABV infection. Abbreviation: 3-MA: 3-methyladenine; ABLV: Australian bat lyssavirus; ATG: autophagy related; AKT: AKT serine/threonine kinase; Baf-A1: bafilomycin A1; CAMKV: CaM kinase like vesicle associated; CAMK2: calcium/calmodulin dependent protein kinase II; co-IP: co-immunoprecipitation; CQ: chloroquine; DUVV: Duvenhage virus; DMSO: dimethyl sulfoxide; EBLV-1: European bat lyssavirus 1; ISG: interferon stimulated gene; LAMP1: lysosome associated membrane protein 1; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; Mdivi-1: mitochondrial division inhibitor-1; MLD₅₀: 50% mouse lethal dose; MOI: multiplicity of infection; MTOR: mechanistic target of rapamycin kinase; qPCR: quantitative real-time polymerase chain reaction; RABV: rabies virus; SQSTM1/p62: sequestosome 1; WT: wild type.
REFERENCE [20] · ID: 42402931
ID: 42402931 Title: Mir452 protects against septic acute kidney injury by targeting Apaf1 to relieve CASP9-mediated suppression of autophagy. Abstract: Septic acute kidney injury (AKI) is associated with high mortality and currently lacks effective therapeutics. Mir452 (microRNA 452) is a newly identified, highly sensitive biomarker for septic AKI, but the biological function of Mir452 was unknown. Here we report that Mir452 protects kidney tubular cells in septic AKI by repressing APAF1 (apoptotic peptidase activating factor 1) and associated caspase activation to preserve macroautophagy/autophagy. Using mouse and cell models of septic AKI induced by lipopolysaccharide (LPS), we found that inhibition of Mir452 exacerbated renal dysfunction, tubular apoptosis, and inflammatory responses, whereas Mir452 mimics significantly attenuated kidney injury. Mechanistically, Mir452 was shown to directly bind to the 3' untranslated region (3'UTR) of Apaf1 mRNA, repressing APAF1 expression and thereby inhibiting apoptosome-mediated CASP9 activation. This repression further alleviated caspase-mediated cleavage of autophagy-related proteins like BECN1 and ATG5, leading to the preservation of autophagic flux, which in turn limits inflammasome activation and inflammation. Notably, tubule-specific deletion of Apaf1 recapitulated the protective effects of Mir452, whereas forced Apaf1 expression aggravated injury, an effect reversed by CASP9 knockdown. Furthermore, Mir452 significantly promotes protective autophagy in septic AKI by suppressing the APAF1-CASP9 axis, as evidenced by upregulated ATG5 and BECN1 expression, enhanced LC3-II accumulation and autophagosome-lysosome fusion, along with reduced SQSTM1/p62 levels. Functional rescue experiments demonstrated that Mir452's anti-inflammatory effects depend entirely on activated autophagy, as overexpression fails when autophagy is inhibited. Together, the results unveil the Mir452-APAF1-CASP9-autophagy signaling axis that provides an intrinsic anti-inflammation and anti-apoptosis mechanism, suggesting new therapeutic targets for septic AKI.
REFERENCE [8] · ID: 42403159
ID: 42403159 Title: Autophagy-related pathway dysregulation and apoptosis suppression in pterygium: Role of key biomarkers Beclin-1, ATG5, LC3, p62, and Bcl-2 in pathogenesis and potential nonsurgical targets. Abstract: ObjectivesTo investigate the roles of apoptosis and autophagy in the pathogenesis of primary pterygium and to evaluate potential biomarkers that could lead to nonsurgical therapeutic approaches.MethodsIn this prospective, observational, controlled study, patients diagnosed with primary pterygium were included. Excised pterygium tissues (study group) were compared with normal conjunctival tissues obtained from autografts (control group). Immunohistochemical staining was used to assess the expression of autophagy-related proteins (Beclin-1, LC3A/B, ATG5, and p62) and apoptosis-related proteins (Bcl-2 and Caspase-8). Quantitative immunohistochemical evaluation was performed using H-score analysis with Image Tool Software.ResultsThe study group exhibited significant increases in the expression of Beclin-1 (97.65 ± 2.54 vs. 25.53 ± 0.4), LC3A/B (97.88 ± 5.35 vs. 61.73 ± 5.08), ATG5 (100.73 ± 1.06 vs. 35.35 ± 0.3), p62 (84.18 ± 3.59 vs. 59.54 ± 2.29), and Bcl-2 (107.36 ± 1.60 vs. 53.56 ± 1.38) compared with the control group (p < 0.001 for all). Although Caspase-8 expression was also increased (65 ± 1.53 vs. 49 ± 0.2), the difference was not statistically significant (p = 0.270). These findings suggest altered regulation of autophagy-related pathways accompanied by suppression of apoptotic mechanisms, which may contribute to the persistence, fibrovascular proliferation, and progression of pterygium tissue.ConclusionThe progression of pterygium appears to be associated with dysregulation of autophagy-related pathways and inhibition of apoptotic mechanisms. These findings suggest that modulation of these cellular mechanisms may provide novel pharmacological therapeutic options, potentially reducing the need for surgical intervention.
REFERENCE [18] · ID: 42404975
ID: 42404975 Title: CCL2 regulates autophagy through the TNF signaling pathway to promote traumatic brain injury progression. Abstract: This study investigated the regulatory role of CCL2 in traumatic brain injury (TBI) and elucidated its underlying molecular mechanism. Bioinformatics analysis revealed significant enrichment of CCL2 in the TNF signaling pathway. ELISA and Western blot analyses confirmed marked upregulation of CCL2 in the serum of TBI patients as well as in the serum and brain tissues of TBI mouse models. Functional experiments demonstrated that CCL2 knockdown significantly alleviated neurological impairment and secondary brain injury in TBI mice. Mechanistically, CCL2 suppression enhanced neuronal autophagy, as evidenced by increased Beclin1 and LC3-II expression and modulation of autophagy flux markers (p62 and LAMP2), while simultaneously attenuating neuronal apoptosis through regulation of apoptosis-related proteins, including Bcl-2, Bax, and cleaved caspase-3. Rescue experiments further showed that TNFR1 overexpression abolished the protective effects of CCL2 knockdown and restored activation of the TNF signaling pathway, accompanied by elevated MCP-1, TNFR1, and phosphorylated p65 levels. Collectively, these findings demonstrate that CCL2 promotes TBI progression by activating the TNF signaling pathway, thereby suppressing autophagy and enhancing apoptosis. Targeting the CCL2-TNFR1 axis may represent a promising therapeutic strategy for secondary brain injury following TBI. The online version contains supplementary material available at 10.1007/s13205-026-04793-0.
REFERENCE [7] · ID: 42405496
ID: 42405496 Title: Zingerone ameliorates ovarian impairment by regulating steroidogenesis and apoptosis in letrozole-treated mouse. Abstract: Polycystic ovary syndrome (PCOS) is characterized by hyperandrogenism, disrupted folliculogenesis, and subfertility. This study evaluated the therapeutic efficacy of zingerone(4-(4-hydroxy-3-methoxyphenyl)-2-butanone) in a letrozole-induced hyperandrogenised PCOS-like mouse model. Zingerone administration, particularly at 25 and 50 mg/kg, significantly improved ovarian morphology by enhancing follicular development and corpus luteum formation. This was accompanied by increased granulosa cell proliferation (PCNA) and attenuation of apoptosis, evidenced by up-regulation of BCL2 and reduced TUNEL staining. Zingerone reprogrammed ovarian steroidogenesis by reducing circulating testosterone, down-regulating androgen receptor expression, suppressing StAR, and up-regulating aromatase, thereby promoting a shift toward estrogen biosynthesis. Autophagy analysis indicated restoration of autophagic flux, reflected by decreased p62 levels and modulation of Beclin1 and LC3B expression. These coordinated molecular and cellular changes resulted in functional recovery, with significant improvement in fertility and litter size at higher doses (25 and 50 mg/kg). Collectively, zingerone exerts dose-dependent, multi-target effects to restore endocrine, cellular, and autophagic homeostasis, thereby ameliorating ovarian dysfunction in PCOS.
REFERENCE [6] · ID: 42405585
ID: 42405585 Title: Targeted Inhibition of RPA3 Impairs Breast Cancer Progression Through Suppressing TGF-β Signaling Pathway-Mediated Autophagy. Abstract: As a key component of the replication protein A (RPA) complex, RPA3 has been identified as oncogenic in multiple solid tumors. However, its specific role in breast cancer remains poorly understood. RPA3 expression and its prognostic relevance in breast cancer were assessed based on the public databases. To further confirm the biological function of RPA3, we knocked down RPA3 in the breast cancer cell line Michigan Cancer Foundation-7 (MCF-7) and then conducted Cell Counting Kit-8, colony formation, Western blot, immunofluorescence, and transmission electron microscopy. In vivo effects of RPA3 were tested in a xenograft model. We found that high expression of RPA3 in breast cancer predicted adverse patient outcomes. RPA3 was mainly involved in multiple oncogenic signaling pathways, including the transforming growth factor-β (TGF-β) pathway. RPA3 knockdown effectively suppressed cancer cell proliferation in vitro and in vivo. Mechanistically, RPA3 knockdown decreased TGF-β1 promoter activity and reduced TGF-β1 expression at mRNA and protein levels, accompanied by decreased p-Smad2/3 levels. RPA3 knockdown also significantly blocked autophagy, as evidenced by decreased microtubule-associated protein 1 light chain 3 (LC3)-II/LC3-I ratio, increased sequestosome 1 (p62) level, and reduced LC3 puncta. Notably, pharmacological activation of the TGF-β pathway partially reversed autophagy alterations induced by RPA3 knockdown. These data support the possibility of RPA3 as a therapeutic target for breast cancer.
REFERENCE [5] · ID: 42406081
ID: 42406081 Title: UBE2L6 promotes invasion and metastasis of triple-negative breast cancer by enhancing autophagy through STK38 ISGylation. Abstract: Although aberrant activation of autophagy is well known in triple-negative breast cancer (TNBC), its functional roles and underlying mechanisms remain largely unknown. In the present study, we found that high UBE2L6 expression was strongly associated with aggressive clinical features in TNBC. We demonstrated that UBE2L6 promoted migration, invasion, and lung metastasis of TNBC by enhancing STK38-mediated autophagy. Mechanistically, UBE2L6 stabilized STK38 by promoting its ISGylation and inhibiting its ubiquitin-proteasomal degradation. Therefore, targeting UBE2L6 and modulating the STK38 ISGylation-autophagy axis represent potential intervention points in TNBC.
REFERENCE [14] · ID: 42409090
ID: 42409090 Title: 3-monochloro-1,2-propanediol disrupts ovarian germline stem cell maintenance via Golgi stress-induced autophagy in Drosophila. Abstract: The ubiquitous food processing contaminant 3-monochloro-1,2-propanediol (3-MCPD) poses a potential threat to female reproductive health, yet the underlying mechanisms remain largely undefined. Here, we demonstrate that 3-MCPD exposure impairs ovarian function by disrupting germline stem cell (GSC) maintenance in Drosophila. 3-MCPD exposure dose-dependently induced ovarian atrophy, reduced fecundity, and GSC loss. Mechanistically, 3-MCPD triggered Golgi stress, leading to Mitf-dependent transcriptional upregulation of Atg9. This, in turn, activated autophagy, which selectively degraded E-cadherin, a critical adhesion molecule for GSC maintenance. Notably, GSC-specific E-cadherin overexpression or Atg9 knockdown effectively rescued 3-MCPD-induced ovarian defects. We further identified a direct physical interaction between Atg9 and E-cadherin, corroborated by immunofluorescence co-localization and AlphaFold3 modeling, which revealed high-affinity binding interfaces that likely mediate this selective autophagic degradation. Both E-cadherin and Atg9 exhibit high evolutionary conservation, underscoring the translational relevance of our findings. This study delineates a novel Golgi stress-Mitf-Atg9 axis through which a prevalent food contaminant compromises female fertility, thereby identifying potential targets for intervention.
REFERENCE [4] · ID: 42409092
ID: 42409092 Title: L-Theanine attenuates isoprenaline-induced heart failure in rats through modulation of the JNK/c-Jun/Beclin 1/Bcl2 pathway. Abstract: Heart failure (HF) is characterized by impaired cardiac function, cardiac hypertrophy, and elevated cardiomyocyte injury biomarkers. Dysregulation of autophagic pathways has been recently implicated in the pathogenesis of HF. The present study aimed to investigate the possible cardioprotective effects of L-theanine in an isoprenaline-induced HF rat model and to study its potential impact on the autophagic process in HF. Forty-two male Wistar rats were randomly allocated into four groups: a normal control group, an HF group (Isoprenaline, 170 mg/kg/day, SC, for 4 days), an HF + L-theanine group (Isoprenaline + L-theanine, 400 mg/kg/day, p.o., for 32 days) and an L-theanine control group. The HF group demonstrated significant deterioration in echocardiographic parameters, accompanied by significant cardiac injury, as evidenced by increased serum LDH and BNP and reduced cardiac troponin I levels. These changes were associated with dysregulated autophagy, as indicated by elevated Beclin-1, LC3-I, LC3-II, and ATG5 expression, along with increased levels of JNK and c-Jun and decreased Bcl-2 levels. Additionally, increased NF-κB levels and altered total antioxidant capacity indicate enhanced inflammatory responses and oxidative stress. Compared with HF, L-theanine administration effectively improved isoprenaline-induced cardiac dysfunction in rats by improving echocardiographic parameters (EF, FS and LVIDs), ameliorating alterations in cardiac injury markers, and attenuating histopathological damage. L-Theanine significantly decreased autophagy as demonstrated by reduced beclin-1, LC3-I, LC3-II, and ATG5 levels. Additionally, L-theanine downregulated p-JNK/c-Jun signaling, reduced NF-κB levels, increased Bcl-2 expression, and increased total antioxidant capacity. This study revealed that the modulation of the JNK/c-Jun/Beclin-1/Bcl-2 pathway by L-theanine ameliorates isoprenaline-induced HF via the regulation of autophagic and inflammatory pathways.
REFERENCE [19] · ID: 42409247
ID: 42409247 Title: CDKN1A protects medium spiny neurons from Huntington's disease pathology. Abstract: Huntington's disease (HD) arises from abnormal expansion of CAG trinucleotide repeats within the HTT gene, leading to mutant huntingtin (mHTT) aggregation, progressive loss of striatal medium spiny neurons (MSNs), and progressive neurodegeneration. While the genetic cause is established, the mechanisms that confer selective MSN vulnerability, particularly those linked to aging, remain unclear. We employed a combination of miR-9/9*-124-driven reprogramming and MSN-specific transcription factors to generate patient-derived MSNs from fibroblasts of symptomatic HD patients (HD-MSNs), pre-symptomatic mutation carriers (pre-HD-MSNs), and healthy controls, preserving donor age signatures. Multi-omics analysis integrating RNA-seq and ATAC-seq revealed reduced CDKN1A expression and promoter accessibility in HD-MSNs compared with pre-HD-MSNs. Overexpression of CDKN1A in HD-MSNs alleviated HD pathologies, including DNA double-strand breaks, oxidative DNA damage, and mHTT aggregates, while improving neuronal survival and autophagy-associated activity. Conversely, knockdown of CDKN1A in pre-HD-MSNs elicited opposite effects, revealing a CDKN1A-dependent survival mechanism in HD. Together, these findings suggest that reduced CDKN1A expression may contribute to HD-associated MSN vulnerability and is associated with altered DNA damage responses and autophagy-related processes in HD-MSNs. Our study identifies CDKN1A as a potential modulator of neuronal resilience in HD.
REFERENCE [3] · ID: 42409251
ID: 42409251 Title: Loss of ETV4 triggers PPM1E downregulation and AMPK-ULK1-dependent autophagy to promote intrinsic trametinib resistance in breast cancer. Abstract: Trametinib, a selective MEK1/2 inhibitor, is approved for melanoma, BRAF-mutant non-small cell lung cancer, and thyroid cancer. Its favorable pharmacologic profile has prompted broader evaluation across cancers driven by MAPK/ERK signaling. However, its efficacy as monotherapy in breast cancer remains limited due to intrinsic resistance. Here, we investigated the molecular basis of intrinsic trametinib resistance and sought strategies to enhance therapeutic response. Trametinib responsiveness was associated with ETV4 expression. Notably, trametinib reduced ETV4 expression in three cell lines with high basal ETV4 expression (MDA-MB-453, SKBR3, and T47D). Consistent with ERK/MAPK-dependent regulation of ETV4, trametinib-mediated MEK inhibition was associated with stabilization of Capicua (CIC), a transcriptional repressor of ETV4, thereby suppressing ETV4 expression. In ETV4-high cells, trametinib-induced ETV4 downregulation promoted autophagic flux. Mechanistically, trametinib treatment and ETV4 silencing induced AMPK Thr172 phosphorylation, leading to ULK1 Ser555 phosphorylation and mTOR inhibition, thereby activating protective autophagy. RNA-seq analysis revealed that trametinib treatment and ETV4 knockdown produced highly overlapping transcriptomic profiles. Notably, trametinib reduced the expression of PPM1E, a phosphatase that negatively regulates AMPK, along with canonical MAPK effector genes. ChIP-PCR analysis and public ChIP-seq data demonstrated that ETV4 directly occupies the PPM1E promoter region and enhances PPM1E transcription, whereas trametinib-induced ETV4 suppression reduced PPM1E expression, limiting AMPK dephosphorylation and thereby promoting AMPK activation. Pharmacological inhibition of autophagy using chloroquine (CQ) or 3-methyladenine (3-MA) enhanced trametinib-induced apoptosis in vitro and suppressed T47D xenograft tumor growth in vivo. Collectively, our findings define a CIC-ETV4-PPM1E-AMPK signaling cascade through which trametinib-induced ETV4 downregulation drives AMPK-ULK1-dependent protective autophagy, thereby conferring a survival advantage in ETV4-high breast cancer. Autophagy blockade restores trametinib sensitivity and induces apoptosis, supporting a combinatorial strategy to improve MEK1/2-targeted therapy.
REFERENCE [17] · ID: 42409767
ID: 42409767 Title: mTORC1 suppression by Trp53 mutation drives resistance to immune checkpoint blockade. Abstract: p53 is a critical tumor suppressor gene that inhibits cancer development by regulating cell cycle arrest, apoptosis, DNA repair, and metabolism. However, recent studies examining TP53 mutations in cancer immunotherapy have yielded inconsistent results, likely due to differences in tumor mutational burden (TMB) and the context-dependent roles of specific p53 mutants. In this study, we assessed the function of G242V and S258I Trp53 mutations in MC38 cells in the context of immunotherapy by generating Trp53 deletion and observed significantly enhanced responses to anti-PD-1 therapy. Trp53-null tumors showed increased CD8+ T cell infiltration and clonal expansion, along with reduced regulatory T (Treg) cells. Mechanistically, Trp53 deletion downregulated mTORC1 inhibitor genes, leading to elevated mTORC1 signaling and diminished autophagy, which sensitized tumor cells to IFN-γ and TNF-α-induced apoptosis. Besides mouse cells, we confirmed the human p53 mutants regulate the same sets of mTORC1 inhibitor genes in a human colorectal cancer cell line. Our findings demonstrate that certain p53 mutants, despite losing other canonical functions, retain wild type p53's ability to suppress mTORC1 and enhance autophagy, thereby inhibiting responses to immunotherapy.
REFERENCE [2] · ID: 42409845
ID: 42409845 Title: Salmonella SopB suppresses post-transcriptionally regulated cytokine release to reduce early tissue inflammation and delay disease progression. Abstract: Salmonella enterica subsp. enterica serovar Typhimurium (S. Typhimurium) manipulates cellular processes through the translocation of effector molecules into the host cell cytosol. Using a recently established neonatal S. Typhimurium infection model, we provide functional insights into how Salmonella outer protein B (SopB) suppresses early mucosal tissue inflammation and prolongs host survival. Mechanistically, SopB prevents a disintegrin and metalloprotease 17 (ADAM17) activation, plasma membrane translocation and the release of membrane-bound TNFα from enterocytes and reduces epithelial secretion of IL-18 via mTOR-controlled secretory autophagy. This abolishes the early epithelial transcriptional response and reduces immune cell recruitment and programmed cell death-mediated mucosal barrier disruption delaying disease progression. The immunosuppressive effect of SopB is independent of the C-terminally encoded phosphatidylinositol phosphatase and phosphotransferase activity but requires an intact N-terminal domain. Also, it is restricted to the neonatal mouse model characterised by Salmonella pathogenicity island (SPI)1 type 3 secretion system (T3SS)-dependent enterocyte invasion-driven mucosal translocation. Thus, here we demonstrate that SopB suppresses the early, post-transcriptional regulation of epithelial cytokine release in an inositol phosphatase-independent manner likely promoting pathogen transmission.
REFERENCE [41] · ID: 42410080
ID: 42410080 Title: SGLT2 inhibition induces autophagic flux blockade and sensitizes pancreatic cancer to EGFR-targeted therapy. Abstract: Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy with profound metabolic rewiring and resistance to therapy. Sodium-glucose cotransporter 2 (SGLT2) regulates glucose uptake, but its role in PDAC remains unclear. SGLT2 expression was analyzed in clinical samples and public datasets. PDAC cell lines were subjected to genetic knockdown or canagliflozin (CANA) treatment to assess proliferation, migration, apoptosis, and glucose metabolism. Mechanistic studies investigated AMPK-ULK1 signaling, autophagy dynamics, oxidative stress, and EGFR signaling. Xenograft models were used to assess in vivo efficacy. SGLT2 was upregulated in PDAC and associated with poor prognosis. SGLT2 inhibition suppressed proliferation and migration while promoting apoptosis. Mechanistically, CANA induced ATP deficiency and initiated autophagy, but concurrently impaired autophagosome-lysosome fusion. This dual effect led to autophagic flux blockade, resulting in excessive ROS accumulation, mitochondrial dysfunction, and apoptosis. Inhibition of AMPK reduced ROS levels, while ROS scavenging partially rescued mitochondrial damage and cell death. Notably, SGLT2 inhibition enhanced sensitivity to EGFR-targeted therapy, producing synergistic anti-tumor effects in vitro and in vivo. SGLT2 maintains metabolic and autophagic homeostasis in PDAC. Its inhibition induces metabolic stress, autophagic flux blockade, and ROS-driven mitochondrial apoptosis. In addition, targeting SGLT2 sensitizes tumors to EGFR-targeted therapy, offering a novel combinatorial strategy.
REFERENCE [32] · ID: 42410256
ID: 42410256 Title: SenFlag gene signature identifies senescent cells in mouse and human tissues through a conserved core transcriptional program. Abstract: Identifying senescent cells via single-cell transcriptome profiling data remains challenging due to cellular heterogeneity and overlap with other cellular states. Here, we present SenFlag, a streamlined gene signature for enhanced identification of senescent cells based on integration of core gene expression features. SenFlag was derived through systematic assessment of bulk and single-cell RNA-sequencing datasets across multiple senescence models. It captures a conserved transcriptional program characterized by reduced expression of proliferation-associated genes and chromatin-associated genes (HMGB1/2, HMGN2), combined with upregulation of cell-cycle inhibitors (CDKN1A/CDKN2A) and of CCND1. Additionally, SenFlag incorporates lysosomal features, including increased expression of V-ATPase subunits and cathepsins. SenFlag identifies a rare but progressively accumulating population of senescent cells across tissues in both mice and humans in vivo, with enrichment in epithelial and endothelial compartments. SenFlag-positive cells increase with age and following tissue injury, and are reduced in datasets involving senescence-targeting interventions, supporting its specificity in vivo. Together, SenFlag provides a robust and interpretable signature for identifying senescent cells in single-cell datasets and facilitates the study of senescence across physiological and pathological contexts.
REFERENCE [31] · ID: 42410284
ID: 42410284 Title: Curcumin Attenuates Cuproptosis via Activating Autophagy Through Inhibition of the AKT/mTOR/P70S6K-Signaling Pathway in Parkinson's Disease Models. Abstract: This research surveyed the therapeutic potential of curcumin (Cur) in Parkinson's disease (PD), focusing on its effects on cuproptosis and underlying molecular mechanisms. A MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine)-induced mouse model and a MPP+ (1-methyl-4-phenylpyridinium)-treated PC12 cell model were used in this study. In vivo, Cur treatment significantly mitigated MPTP-treated dyskinesia and lessened the damage of dopaminergic (DA) neurons in SNpc. Additionally, Cur reversed MPTP-induced changes by increasing TH (tyrosine hydroxylase) expression and decreasing α-syn (α-synuclein) accumulation in the SN. In vitro, Cur mitigated MPP+-treated apoptosis and the cytotoxicity of differentiated PC12 cells. Furthermore, Cur reversed MPTP/MPP+-induced changes in the cuproptosis-related protein expression, including DLAT (dihydrolipoamide S-acetyltransferase), FDX1 (ferredoxin 1), and upregulating SLC31A1 (solute carrier family 31 member 1) and HSP70 (heat shock protein 70). 3-MA (3-methyladenine) reversed Cur-mediated expression levels of DLAT, FDX1, SLC31A1, and HSP70 in the PD models. Mechanistically, Cur decreased the expression of p-AKT (p-protein kinase B), p-mTOR (p-mammalian target of rapamycin), and p-P70S6K (p-70 KDa ribosomal protein S6 kinase) in the PD models, suggesting it has an inhibitory effect on the AKT/mTOR/P70S6K signaling pathway. Furthermore, pretreatment with SC79 (an AKT activator) reversed Cur-induced autophagy activation, supporting the role of this pathway in Cur-mediated neuroprotection. Cur protected against DA neuronal loss by modulating the interplay between cuproptosis and autophagy via the suppression of the AKT/mTOR/P70S6K. The study findings provide novel insights into the mechanism of Cur's neuroprotective effect, highlighting the AKT/mTOR/autophagy/cuproptosis axis as a potential target and Cur as a medicant for PD management.
REFERENCE [30] · ID: 42410294
ID: 42410294 Title: Shared genetic liability between opioid analgesic use and lung cancer subtypes revealed by genome wide correlation and structural modeling. Abstract: Observational studies link opioid use to lung cancer risk, but findings are inconsistent due to potential confounding and reverse causality. Whether these associations reflect shared genetic liability with histologic lung cancer (LC) subtypes is unknown. This study quantified genome-wide genetic overlap and modeled their latent shared architecture. This analysis used European-ancestry genome-wide association study (GWAS) summary statistics for opioid use traits (codeine/tramadol and dihydrocodeine) and lung cancer outcomes (overall lung cancer, non-small cell lung cancer, adenocarcinoma, and squamous cell carcinoma). Bivariate linkage disequilibrium score regression (LDSC) estimated heritability and genetic correlations. Genomic structural equation modeling (Genomic SEM) tested latent factor models, and multi-marker analysis of genomic annotation (MAGMA) performed gene, pathway, and tissue enrichment analyses. An exploratory Mendelian randomization (MR) analysis was additionally conducted when adequate instrumental variants were available. LDSC indicated uniformly positive genetic correlations between opioid traits and lung cancer outcomes, strongest for CT-NSCLC (rg = 1.1017, p = 6.86 × 10- 4) and DHC-NSCLC (rg = 0.9773, p = 4.46 × 10- 2); other positive pairs included DHC-LC (rg = 0.5938, p = 1.59 × 10- 5) and DHC-SCC-L (rg = 0.6366, p = 9.03 × 10- 4), with remaining correlations smaller but positive (CT-LC rg = 0.2702; CT-LAC rg = 0.2055; CT-SCC-L rg = 0.3358; DHC-LAC rg = 0.3377). Genomic SEM supported a two-factor model (CFI > 0.99; SRMR = 0.0602) separating cancer outcomes (LAC β = 0.64, LC β = 1.10, SCC-L β = 0.83) from opioid traits (DHC β = 1.22; CT β = 0.61). MAGMA identified enrichment for oxidative phosphorylation/mitochondrial electron transport chain, Notch, cell-cycle, DNA damage response-p53/TP53, and immune pathways. Exploratory MR was feasible only for CT under the prespecified instrument-selection criteria, whereas DHC did not yield sufficient instruments and therefore could not be evaluated by MR. The CT-based MR analysis did not provide robust evidence for a causal effect of codeine/tramadol use liability on lung cancer outcomes, indicating that the observed LDSC associations are more appropriately interpreted as shared genetic liability rather than confirmed causality. Common-variant liability is broadly shared between opioid medication use and lung cancer, particularly CT-NSCLC, with a correlated two-factor structure separating cancer susceptibility from medication use. Enrichment analyses highlighted mitochondrial energetics, DNA damage response/TP53, immune signaling, and subtype-specific pathways. Exploratory MR was feasible only for CT and did not support a definitive causal interpretation, reinforcing the need to view the findings primarily as evidence of shared genetic liability.
REFERENCE [26] · ID: 42410873
ID: 42410873 Title: Comparative Biophysical Analysis of Healthy and Inflamed Intestinal Membrane Models Using Langmuir Monolayers. Abstract: Intestinal inflammation, including Crohn's disease, represents a chronic condition that increases the risk of colon cancer and involves complications from long-term pharmacotherapy. Since drugs must pass through lipid bilayers to reach their targets, understanding the surface properties of biological membranes is crucial. In this study, the Langmuir technique and Brewster angle microscopy (BAM) were employed to characterize simplified models of healthy and inflamed intestinal cell membranes. Both the outer and inner leaflets of the lipid bilayer were modeled to investigate how inflammation-induced changes in lipid composition, fatty acid saturation, and pH affect membrane integrity. The results demonstrate that inflamed models exhibit increased fluidity, reduced molecular packing, and lower collapse pressures compared to healthy ones. Notably, the study reveals a significant reduction in the differentiation between the surface properties of the outer and inner monolayers in the inflamed state, indicating a loss of membrane asymmetry. These findings provide novel physicochemical insights into inflammation-driven membrane remodeling, potentially supporting the design of targeted therapies with improved absorption in pathological states.
REFERENCE [25] · ID: 42410910
ID: 42410910 Title: Targeting the SNAI1-LAMP3 axis to restore lysosomal function and alleviate autophagic flux impairment to delay retinal degeneration. Abstract: Retinal degenerative diseases are a leading cause of irreversible blindness. Their pathogenesis is intricately linked to oxidative stress-induced dysfunction of retinal pigment epithelial (RPE) cells and subsequent retinal degeneration. Macroautophagy/autophagy, a critical cellular degradation pathway, plays a vital role in maintaining RPE homeostasis, yet its dysregulation in retinal degenerative diseases remains poorly understood. In this study, we observed that sodium iodate (NaIO3), an oxidative stress inducer, triggered lysosomal dysfunction via lysosomal membrane permeabilization (LMP), thereby impairing autophagic flux in RPE cells and exacerbating retinal degeneration. RNA sequencing identified Lamp3 (lysosomal-associated membrane protein 3) as a downregulated gene following NaIO3 treatment. Functionally, LAMP3 overexpression alleviated NaIO3-induced LMP, improved lysosomal function, and alleviated autophagic impairment. Furthermore, upregulation of LAMP3 reduced oxidative stress and apoptosis in RPE cells, while alleviating retinal degeneration in a NaIO3-induced mouse model. Mechanistically, our data suggested that NaIO3 upregulated the transcription factor SNAI1, which acts as a transcriptional repressor of LAMP3. SNAI1 knockdown increased LAMP3 expression, thereby facilitating the recovery of lysosomal function and the alleviation of autophagic impairment. Collectively, our findings indicate that the SNAI1-LAMP3 axis contributes to the regulation of the autophagy-lysosomal pathway in retinal degeneration, highlighting a potential therapeutic target for delaying disease progression.
REFERENCE [15] · ID: 42410967
ID: 42410967 Title: Targeting lysosomes with a novel chloroquine derivative induces irreversible lysosomal damage and disrupts autophagosome and lysosome assembly in cancer. Abstract: Pancreatic ductal adenocarcinoma (PDAC) exhibits profound therapy resistance driven by lysosome-dependent nutrient recycling, metabolic adaptation, and stress tolerance. Current lysosome targeting agents such as chloroquine (CQ)/hydroxychloroquine (HCQ) show limited efficacy due to transient activity and dose-limiting-toxicities. To overcome these limitations, we developed lysostilbenes, a new class of hybrid small molecules combining the CQ pharmacophore with lysosome-disrupting stilbene analogs. Stilbene pharmacophore is the core structural component of resveratrol. Among the synthesized hybrids, lysostilbene-4 emerged as the lead candidate, demonstrating ~30-40-fold greater cytotoxicity against PDAC cells than parent compounds, while sparing nonmalignant cells. At nanomolar concentrations, lysostilbene-4 induced rapid, irreversible lysosomal membrane permeabilization (LMP), initiating a lysosome mitochondria apoptotic cascade via CTSB (cathepsin B) release, BID cleavage, BAX activation, and caspase-mediated apoptosis. In parallel, it abrogated lysosomal recovery by significantly reducing repair, lysophagy, autophagosome maturation, and uncoupling TFEB-driven transcriptional programs from effective lysosome biogenesis. Reduced TFEB mRNA expression correlated with poor overall-survival and disease-free-survival across multiple cancer patients, with a particularly strong association in pancreatic cancer patients. Using TFEB+/+ and TFEB-/- knockout pancreatic cancer cells we establish that lysostilbene-4 exerts severe cytotoxicity by inducing persistent lysosomal-damage and disrupting autophagosome-lysosome assembly, with vulnerability further amplified in TFEB-deficient cells. This finding underscores TFEB as a key determinant of lysosomal-resilience and a potential predictive biomarker. Importantly, lysostilbene-4 was well tolerated in preclinical mouse-models at supra-therapeutic doses without systemic-toxicity. These findings position lysostilbene-4 as a first-in-class lysosome-targeting therapeutic that enforces sustained lysosomal collapse while compromising adaptive recovery-mechanisms, providing a mechanistically precise and safe strategy against PDAC.Abbreviations: ALG: autophagy-lysosome genes; AMPK: AMP-activated protein kinase; CASM: conjugation of ATG8s to single membranes; CTSB: cathepsin B; LGALS3: galectin 3; LMP: lysosomal membrane permeabilization; LS: lysostilbene; MTOR: mechanistic target of rapamycin kinase; PDAC: pancreatic ductal adenocarcinoma; TCGA: The Cancer Genome Atlas; TFEB: transcription factor EB; ULK1: unc-51 like autophagy activating kinase 1.
REFERENCE [37] · ID: 42411040
ID: 42411040 Title: Thrombospondin-4 Regulates Lipopolysaccharide-Induced Apoptosis and Inflammation in Nucleus Pulposus Cells via the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway. Abstract: This study investigated the expression profile of Thrombospondin-4 (THBS4) in intervertebral disc degeneration (IDD) and clarify its regulatory role in lipopolysaccharide (LPS)-induced apoptosis and inflammation in nucleus pulposus cells (NPCs) via the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) pathway. IDD-related differentially expressed genes were screened through the integration of GeneCards and the Gene Expression Omnibus (GEO) database (GSE186542), followed by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis to explore potential signaling mechanisms. An in vitro inflammatory injury model was established using LPS-stimulated NPCs. The effects of THBS4 overexpression on cell proliferation, apoptosis, inflammatory cytokine secretion, and extracellular matrix protein expression were evaluated using reverse transcription-quantitative polymerase chain reaction (RT-qPCR), western blotting, 5-ethynyl-2'-deoxyuridine (EdU) assay, flow cytometry, and enzyme-linked immunosorbent assay (ELISA). Additionally, the involvement of the PI3K/AKT pathway in mediating THBS4-related effects was confirmed using the PI3K inhibitor LY294002. Bioinformatics analysis revealed that THBS4 was significantly downregulated in IDD and was closely linked to the PI3K/AKT pathway. Functional assays demonstrated that overexpression of THBS4 markedly enhanced NPCs proliferation, suppressed apoptosis, reduced the secretion of tumour necrosis factor alpha (TNF-α), interleukin-1beta (IL-1β) and IL-6, and increased the expression of IL-10, Aggrecan, and Collagen type II. These protective effects were accompanied by activation of the PI3K/AKT pathway and were significantly reversed by LY294002 treatment. THBS4 alleviated LPS-induced damage in NPCs through the activation of the PI3K/AKT pathway, exerting anti-apoptotic and anti-inflammatory effects; the specific upstream molecular mechanism of PI3K/AKT activation by THBS4 requires further investigation. These findings suggested that THBS4 may serve as a potential therapeutic target for IDD treatment.
REFERENCE [44] · ID: 42411048
ID: 42411048 Title: LncRNA Mirt2 Attenuates Osteoarthritis Progression by Promoting miR-429/TBK1-Mediated Autophagy. Abstract: Osteoarthritis (OA) is a chronic and degenerative joint disorder that is prevalent in middle-aged and older populations. While several long non-coding RNAs (lncRNAs) have been implicated in OA progression, the role of lncRNA Mirt2 and its regulatory mechanisms remain unclear. The expression of lncRNA Mirt2, miR-429, and TANK-binding kinase 1 (TBK1) was detected using qRT-PCR in normal and OA cartilage tissues. The interleukin (IL)-1β-stimulated chondrocyte was used as an in vitro OA cell. EdU, TUNEL assay, western blot, and ELISA assays were used for our experiments. Luciferase reporter assays, RNA immunoprecipitation (RIP), and RNA pulldown were used to investigate the interactions between lncRNA Mirt2, miR-429, and TBK1. An in vivo OA model was established, and cartilage damage was evaluated using H/E and Safranin O/Fast Green staining. LncRNA Mirt2 expression was significantly downregulated in OA cartilage tissues and IL-1β-stimulated chondrocytes (p < 0.05). Transfection of the lncRNA Mirt2 overexpression vector led to a remarkable increase in cell proliferation, a significant reduction in cell apoptosis and inflammation, and a marked elevation in autophagy in IL-1β-induced chondrocytes (p < 0.05). Functional investigation revealed that lncRNA Mirt2 acts as a competing endogenous RNA (ceRNA) by sponging miR-429 in IL-1β-induced chondrocytes. Additionally, miR-429 directly targets TBK1. Rescue experiments showed that overexpression of miR-429 or inhibition of TBK1 effectively counteracted the functional consequences of lncRNA Mirt2 upregulation in IL-1β-stimulated chondrocytes, reversing its promotive effects on cell proliferation and autophagy, as well as its inhibitory effect on apoptosis. The in vivo experiments indicated overexpression of lncRNA Mirt2 down-regulated miR-429, up-regulated TBK1, substantially attenuated cartilage damage, and accelerated autophagy in OA mice (p < 0.05). LncRNA Mirt2 promoted chondrocyte proliferation and alleviated chondrocyte apoptosis, inflammation, and cartilage degeneration by activating miR-429/TBK1-mediated autophagy. Consequently, lncRNA Mirt2 could be a potential target for OA diagnosis and treatment.
REFERENCE [29] · ID: 42411331
ID: 42411331 Title: Targeting Ferroptosis: Effects and Mechanisms of Action of Berberine. Abstract: Ferroptosis, a form of regulated cell death characterized by iron-dependent lipid peroxidation, plays a critical role in various diseases. Berberine, a bioactive compound from plants such as Coptis chinensis, tree turmeric, and barberry, exhibits bidirectional regulation of ferroptosis, but its systemic mechanisms remain unclear. This review summarizes berberine's effects through multiple signaling pathways, emphasizing context-dependent mechanisms, tissue-specific accumulation, and therapeutic potential. Relevant literature was retrieved from PubMed, Web of Science, CNKI, and ScienceDirect. Berberine modulates ferroptosis via iron homeostasis, lipid peroxidation, the System Xc-/GSH/GPX4 anti-oxidant system, and mitochondrial function, involving NRF2, p53, AMPK, PI3K/Akt, and MAPK pathways. It promotes ferroptosis in tumors and fibrotic diseases but inhibits it in cardiovascular, metabolic, and neurological disorders, alleviating tissue damage. Nano-delivery systems and structural optimization enhance bioavailability and targeting, demonstrating therapeutic efficacy and biosafety. Berberine's multi-target, bidirectional regulation shows promising clinical potential, warranting further mechanistic and delivery-focused studies.
REFERENCE [38] · ID: 42411410
ID: 42411410 Title: Neurochondrin promotes U5 snRNP maturation by regulating AAR2 release from PRPF8. Abstract: Pre-mRNA splicing is orchestrated by the spliceosome, a dynamic and highly regulated ribonucleoprotein complex composed of five small nuclear ribonucleoproteins (snRNPs). Despite extensive studies, the biogenesis of snRNPs remains incompletely understood. Here, we identify neurochondrin (NCDN) as a critical regulator of U5 snRNP biogenesis. NCDN associates with PRPF8-AAR2-EFTUD2 complex in the cytoplasm and is essential for the proper progression of this assembly intermediate toward mature U5 snRNP formation. Loss of NCDN causes the accumulation of this intermediate, resulting in a decreased level of mature U5 snRNP. Spliceosome dysregulation often leads to alternative splicing abnormalities implicated in cancer. Indeed, NCDN deficiency suppresses tumor cell proliferation and induces apoptosis, while high NCDN expression promotes tumor cell growth and correlates with poor survival in glioblastoma patients. Transcriptome analyses reveal that loss of NCDN causes widespread alternative splicing defects and changes in gene expression. Collectively, these results establish NCDN as an essential factor for U5 snRNP assembly and spliceosome function, and highlight its potential as a therapeutic target in glioma with elevated NCDN expression.
REFERENCE [24] · ID: 42411475
ID: 42411475 Title: Broad Roles of Endoplasmic Reticulum Stress Sensors Activated by Diverse Pathophysiological Stimuli. Abstract: The endoplasmic reticulum (ER) stress response is a critical cellular program that maintains proteostasis and membrane homeostasis through the activation of the ER stress sensor proteins inositol-requiring enzyme 1 (IRE1), protein kinase R-like ER kinase (PERK), activating transcription factor 6 (ATF6), and old astrocyte specifically induced substance (OASIS) family proteins. These sensors, canonically understood as transducers of the unfolded protein response (UPR), respond to the accumulation of misfolded proteins in the ER lumen as a result of ER luminal Ca2+ depletion, defective disulfide bond formation, dysregulated glycosylation, or inhibition of ER-associated degradation. However, recent conceptual advances have reshaped understanding of these classical mechanisms, by revealing multiple non-canonical pathways that operate independently of luminal proteotoxicity. Emerging evidence highlights the roles of ER stress sensors in integrating diverse stimuli, including the integrated stress response, lipid bilayer stress, mitochondria-ER contact, and the DNA damage response. Herein, we discuss how these ER stress sensors function as multidimensional signaling hubs for proteotoxic, metabolic, and genomic stresses, and consequently modulate pathophysiological cellular outcomes. Finally, we examine current knowledge regarding both canonical and non-canonical modes of ER stress sensor activation, and we discuss how these mechanisms expand the functional scope of ER stress signaling in physiological regulation and diseases.
REFERENCE [35] · ID: 42411477
ID: 42411477 Title: Six Dehydrogenase Gatekeepers of Carbohydrate Metabolism: Metabolic Integration in Health and Disease. Abstract: Dehydrogenases function as metabolic gatekeepers, regulating carbon flux, redox balance, and biosynthetic capacity at critical branch points in cellular metabolism. This narrative review examines six key dehydrogenases, namely glyceraldehyde-3-phosphate dehydrogenase (GAPDH), lactate dehydrogenase (LDH), pyruvate dehydrogenase complex (PDHC), malate dehydrogenase (MDH1/2), isocitrate dehydrogenase (IDH1/2/3), and glucose-6-phosphate dehydrogenase (G6PDH), that collectively orchestrate the partitioning of nutrients among energy production, biosynthesis, and redox homeostasis. These enzymes share common features, including cofactor-dependent catalysis (NAD+/NADH or NADP+/NADPH), strategic positioning at metabolic nodes, and integration of compartmentalized metabolism between the cytosol and mitochondria. Under physiologic conditions, these dehydrogenases enable metabolic flexibility, allowing cells to adapt nutrient utilization to changing energetic demands and biosynthetic requirements. However, their dysregulation drives pathogenesis across diverse human diseases. In cancer, altered dehydrogenase activity supports metabolic reprogramming, exemplified by the Warburg effect mediated by LDHA, oncometabolite production (mutant IDH1/2), and enhanced biosynthetic capacity associated with G6PDH activity. Metabolic syndrome and diabetes feature PDHC suppression via pyruvate dehydrogenase kinase (PDK) upregulation, contributing to metabolic inflexibility and impaired glucose oxidation. Inherited enzymopathies, including G6PDH and PDHC deficiencies, underscore the essential roles of these enzymes and their tissue-specific requirements. In neurodegenerative disorders, oxidative modification of GAPDH promotes protein aggregation, whereas age-related decline in NAD+ compromises the activity of multiple NAD+-dependent dehydrogenases in a tissue- and context-dependent manner. The central importance of these enzymes has generated substantial therapeutic interest. Successful clinical translation includes mutant IDH inhibitors that reverse oncometabolite-driven epigenetic reprogramming in cancer. However, targeting essential metabolic enzymes presents challenges, including narrow therapeutic windows, metabolic compensation, and tissue-specific toxicities. Future therapeutic strategies will likely focus on exploiting disease-specific vulnerabilities, developing isoform-selective inhibitors, and combining metabolic interventions with conventional therapies. Understanding these six dehydrogenase gatekeepers provides crucial insights into metabolic regulation and highlights opportunities for precision-medicine approaches targeting the metabolic dependencies of human disease.
REFERENCE [33] · ID: 42411498
ID: 42411498 Title: Navigating the Redox Precipice: Metabolic Gatekeeping as a Therapeutic Window in Pancreatic Precancer. Abstract: Pancreatic ductal adenocarcinoma (PDAC) remains a lethal malignancy largely because its early stages evade detection. Two recent studies by Hennequart et al. and Radyk et al. (2026, Nature Metabolism) agree on the following concept: pancreatic acinar cells must maintain a narrow "redox precipice" during acinar-to-ductal metaplasia (ADM), the initial reversible precursor of PDAC. Redundant nicotinamide adenine dinucleotide phosphate-generating systems-mitochondrial aldehyde dehydrogenase 1 family member L2 and cytosolic glucose-6-phosphate dehydrogenase/malic enzyme 1-generally regulate reactive oxygen species within a range that facilitates pro-survival signaling without inducing cell death. Disruption of these systems promotes ADM formation and pancreatic intraepithelial neoplasia, whereas antioxidant treatment inhibits progression. This Opinion integrates these findings within a broader framework of metabolic gatekeeping, discusses how the redox precipice interacts with epigenetic reprogramming and immune evasion, and proposes that the transition from redox balance to addiction results in stage-specific vulnerabilities. Circulating formate emerges as a promising biomarker for early detection, and we highlight key unanswered questions, including whether similar principles apply to other metaplasia-driven malignancies.
REFERENCE [28] · ID: 42411514
ID: 42411514 Title: Electroacupuncture Alleviates Focal Cerebral Ischemia-Reperfusion Injury and Is Associated With Modulation of Autophagy-Ferroptosis Involving the STAT3/HIF-1α Signalling Pathway. Abstract: Focal cerebral ischemia-reperfusion injury remains a major clinical challenge in stroke management. Electroacupuncture (EA) may confer neuroprotection by modulating key cellular processes; however, its precise role in regulating autophagy-ferroptosis crosstalk remains largely unclear. The present study aimed to investigate the neuroprotective potential of EA in cerebral ischemia-reperfusion injury, with a focus on exploring potential pathways involving autophagy and ferroptosis regulation. Focal cerebral ischemia-reperfusion injury was modelled using middle cerebral artery occlusion/reperfusion (MCAO/R) in vivo and oxygen-glucose deprivation/reperfusion (OGD/R) in vitro. The therapeutic effect of EA on MCAO/R mice was assessed using several methods, including behavioural tests, cerebral blood flow measurement, and cerebral infarction volume analysis. Molecular analyses used immunofluorescence staining, western blot analysis, and transmission electron microscopy to examine signal transducer and activator of transcription 3 (STAT3)/hypoxia-inducible factor-1α (HIF-1α) pathway activity and its relationship with autophagy/ferroptosis markers. In MCAO/R mice, EA intervention improved neurological functional recovery, decreased cerebral infarction, and enhanced blood flow. EA also downregulated activation of the STAT3/HIF-1α signalling pathway. Furthermore, EA was associated with reduced markers of excessive autophagy and reduced ferroptosis markers in neurons. IL-6-mediated enhancement of STAT3 phosphorylation significantly weakened EA's protective effects against cerebral ischemia-reperfusion injury. In vitro, STAT3 knockdown prevented OGD/R-induced activation of STAT3/HIF-1α signalling and was accompanied by reduced autophagy and ferroptosis markers. Our results suggest that EA exerts neuroprotective effects against cerebral ischemic injury, which may be associated with modulation of autophagy and ferroptosis markers via the STAT3/HIF-1α signalling pathway.
REFERENCE [34] · ID: 42411668
ID: 42411668 Title: The Dual Role of Non-Coding RNAs in Regulating HSC and MSC Fate: Modulating Survival, Death, and Intercellular Communication. Abstract: Non-coding RNAs (ncRNAs) play crucial roles in regulating hematopoietic and mesenchymal stem cells skewing towards regenerative or modulating commitment. There is gap in understanding how ncRNAs regulate diverse pathways that offer new opportunities for therapeutic targeting in regenerative medicine and disease management. The current review examines the dual regulatory mechanisms of ncRNAs in stem cell biology, analyzing their roles as positive (upscale) and negative (downscale) cellular modulators through comprehensive review of ncRNA signaling pathways. As positive regulators, ncRNAs promote cell survival, regulate the cell cycle, enhance differentiation, selfrenewal, delay senescence, and modulate autophagy to maintain stem cell function. As negative regulators, ncRNAs induce various forms of cell death, including apoptosis, necroptosis, pyroptosis, and ferroptosis, often through interactions with long ncRNAs. Additionally, ncRNAs modulate inflammatory responses by influencing proinflammatory cytokines and reducing cell adhesion, further impacting stem cell survival. ncRNAs also influence intercellular communication and signaling pathways, enhancing or suppressing cellular crosstalk essential for stem cell differentiation. Therefore, ncRNAs demonstrate complex dual regulatory functions in stem cell biology, serving both as protective and detrimental influencers/modulators depending on cellular context. Future research should focus on elucidating ncRNA signaling networks and developing ncRNA-based interventions for stem cell dysfunction and associated pathologies.
REFERENCE [23] · ID: 42411996
ID: 42411996 Title: Discovery of Effective Dual PROTAC Degraders with Synergistic Antitumor Activity for Overcoming Tamoxifen-Resistant Breast Cancer. Abstract: Endocrine therapies for estrogen receptor-positive (ER+) breast cancer (BC) often encounter primary or acquired resistance, and elevated expression of histone deacetylase 6 (HDAC6) exacerbates therapeutic challenges. Emerging evidence indicates that simultaneous attenuation of estrogen receptor α (ERα) and HDAC6 activities represents a promising strategy to overcome endocrine resistance. Herein, we report a novel hybrid ERα/HDAC6 dual-targeting PROTAC V-12c, which selectively degraded ERα and HDAC6 in vitro and in vivo and exhibited superior antiproliferation efficacy across a panel of BC cell lines. Mechanistically, V-12c degraded both ERα and HDAC6 via the proteasome pathway, induced cell cycle arrest and apoptosis, attenuated hormonal response, disrupted the autophagy-lysosome function, and triggered ferroptosis, collectively contributing to overcoming tamoxifen resistance. Importantly, V-12c potently suppressed tumor growth in endocrine-resistant BC models, outperforming single-target therapies. This study identified a multimechanistic action of dual PROTAC V-12c with potent antitumor effects, providing a promising therapeutic strategy for endocrine-resistant BC.
REFERENCE [36] · ID: 42412246
ID: 42412246 Title: Endosymbiotic theory of aging revisited: Age-related leakage of mitochondrial dsDNA/RNA stimulates cytosolic nucleic acid sensors which remodel the immune network and promote the aging process. Abstract: About 1.5-2 billion years ago, an endosymbiosis between aerobic α-proteobacteria and anaerobic archaeal cells generated mitochondria, i.e., organelles capable of producing oxidative energy. The bacterial genome was fundamentally reduced and a circular mitochondrial genome evolved containing mainly the genes coding for the subunits of the electron transport chain. Before the symbiotic event, there existed a virus-host co-evolution which involved the development of sensors for detecting dangerous viral DNA/RNA molecules. Endosymbiosis supplied eukaryotic cells not only with an oxidative powerhouse to allow the evolution of more complex multicellular organisms but it also meant that cells now housed an organelle which was able to generate reactive oxygen species (ROS) and to leak mitochondrial DNA (mtDNA) and double-stranded RNA (dsRNA) into the cytoplasm. There is now abundant evidence that during aging and age-related diseases mitochondria are prone to release both mtDNA and dsRNA. In the cytoplasm, mtDNA/dsRNA molecules activate a number of cytosolic nucleic acid sensors leading to the secretion of type-1 interferons (IFN) and many other cytokines which promote an age-related proinflammatory state. Currently, it is known that mtDNA can activate the cGAS-STING pathway, AIM2 inflammasomes, IFI16 receptors, and ZBP1 sensors and in addition mitochondrial dsRNA stimulates RIG-1/MDA5 signaling. Interestingly, there is abundant evidence that all these receptors are drivers of cellular senescence and inflammaging. For decades, there has been mounting evidence that mitochondria have a crucial role in the aging process. We will examine this question from the perspective of evolution and propose that mitochondrial evolution created an endogenic source for the leakage of dangerous mtDNA/dsRNA which subsequently stimulated cytosolic DNA/RNA sensors, an evolutionarily conserved viral defence mechanism. It seems that these two evolutionary events provided not only the basis for the inevitable process of aging but also ensuring the death of parental organisms.
REFERENCE [1] · ID: 42412300
ID: 42412300 Title: Dehydrocostus Lactone Activates Nrf2 Signaling Pathway to Attenuate Oxidative Stress, Inflammation, and Excessive Autophagy in a Mouse Model of Chronic Obstructive Pulmonary Disease. Abstract: Chronic obstructive pulmonary disease (COPD) is a progressive and debilitating respiratory disorder associated with high global mortality. Dehydrocostus lactone (DHLC), a natural sesquiterpene lactone derived from Saussurea lappa Clarke (a medicinal plant), possesses documented antioxidant and anti-inflammatory properties. This study aimed to investigate the potential therapeutic role and mechanism of action of DHLC in COPD. For in vivo experiments, male wild-type and Nrf2-knockout C57BL/6J mice were randomized into control, cigarette smoke extract (CSE), and CSE + DHLC treatment groups. Pulmonary function was assessed by measuring airway resistance and dynamic compliance Histopathological changes were assessed by hematoxylin and eosin staining, and emphysema severity was quantified by mean linear intercept and mean alveolar area measurements. Matrix metalloproteinase-9 (MMP-9) expression was detected by immunofluorescence, while oxidative stress markers superoxide dismutase (SOD) activity and malondialdehyde (MDA) level were measured using commercial kits. For in vitro experiments, mouse alveolar epithelial MLE-12 cells were cultured and exposed to 5% CSE for 24 h. Cell viability was determined by CCK-8 assay, intracellular ROS generation was detected using the DCFH-DA probe, and inflammatory cytokine levels in cell supernatants and BALF were quantified by ELISA. Protein expression of Nrf2, HO-1, LC3, p62, and other targets was analyzed by western blotting; Nrf2 subcellular localization was visualized by immunofluorescence staining; and mRNA expression was measured by RT-qPCR. DHLC significantly improved pulmonary function, alleviated inflammatory cell infiltration and pulmonary emphysema, and reduced MMP-9 expression in the lungs of COPD mice. In both CSE-induced MLE-12 cells and murine COPD models, DHLC attenuated inflammation, oxidative stress, and excessive autophagy by decreasing pro-inflammatory factor levels, ROS generation, the LC3-II/I ratio, and MDA content, while increasing p62 expression and SOD activity. Furthermore, DHLC up-regulated Nrf2 and HO-1 expression and promoted Nrf2 nuclear translocation in CSE-exposed models. Most importantly, siRNA-mediated knockdown of Nrf2 abolished the protective effects of DHLC against CSE-induced inflammation, oxidative stress, and dysregulated autophagy. DHLC ameliorates CSE-induced COPD-like pathology in mice by attenuating oxidative stress, inflammation, and excessive autophagy through activation of the Nrf2 pathway.
REFERENCE [22] · ID: 42412302
ID: 42412302 Title: Study on the Effects and Mechanisms of Resveratrol in Improving Cognitive Impairment in Aβ1-42-Induced Alzheimer's Disease Model Mice. Abstract: Aging is a major risk factor for neurodegenerative diseases, including Alzheimer's disease (AD). Targeting cellular senescence has therefore emerged as a promising therapeutic strategy. Resveratrol (RV), a natural polyphenolic compound, exhibits anti-aging properties through the regulation of autophagy and oxidative stress; however, its mechanisms in AD remain incompletely understood. In this study, we investigated the effects and underlying mechanisms of RV in an Aβ1-42-induced AD model. In vivo, RV administration significantly reduced the expression of aging-related markers and activated autophagy-associated signaling pathways. In vitro, RV treatment markedly attenuated Aβ1-42-induced cell viability loss and excessive reactive oxygen species (ROS) production. Further mechanistic analyses demonstrated that RV-induced autophagy activation was closely associated with the AMP-activated protein kinase/UNC-51-like kinase 1 (AMPK/ULK1) and silent information regulator 1/nuclear factor-kappaB (SIRT1/NF-κB) pathways. Collectively, these findings suggest that RV alleviates AD-related pathological processes by promoting autophagy and delaying cellular senescence, highlighting its potential as a therapeutic agent for age-related neurodegenerative diseases.
REFERENCE [21] · ID: 42412329
ID: 42412329 Title: Mitophagy in Metabolic Dysfunction-Associated Fatty Liver Disease: Mechanisms, Regulatory Networks, and Therapeutic Perspectives. Abstract: Metabolic dysfunction-associated fatty liver disease (MASLD) represents the most prevalent chronic liver disorder globally, with pathogenesis closely linked to insulin resistance, obesity, and gut microbiota dysbiosis. Mitochondrial dysfunction is central to MASLD progression, and mitophagy-a selective form of autophagy that clears damaged mitochondria-plays a crucial role in maintaining cellular homeostasis. This review systematically delineates the molecular mechanisms, regulatory networks, and therapeutic implications of mitophagy in MASLD. We first outline the core machinery of mitophagy, encompassing both ubiquitin-dependent and ubiquitin-independent pathways. We then discuss how impaired mitophagy drives the disease progression of MASLD from the perspective of different hepatic cell types. Furthermore, we summarize the multilayered upstream regulatory network governing mitophagy in the context of MASLD, involving key signaling pathways, metabolic reprogramming, inflammatory cues, epigenetic modifications, and intercellular crosstalk. Finally, we examine therapeutic strategies targeting mitophagy-including clinical and preclinical agents, natural compounds, physical interventions, and emerging technologies-and highlight the challenges posed by its dualistic nature. Moving forward, integrating spatiotemporal dynamics with precision targeting will be essential to translate mitophagy modulation from mechanistic insight into viable clinical therapies for MASLD.
REFERENCE [27] · ID: 42412383
ID: 42412383 Title: Epigenetic mechanisms of gingival aging: biological basis, periodontal implications, and therapeutic perspectives. Abstract: Gingival aging has become increasingly important as increasing number of individuals retain their natural dentition into older age. Beyond epithelial thinning, connective tissue remodeling, vascular alterations, and delayed healing, aging gingiva may be affected by immunosenescence, inflammaging, cellular senescence, and epigenetic dysregulation. Epigenetic mechanisms, including DNA methylation, histone modification, and microRNA-mediated regulation, may link chronological aging and cumulative environmental exposures to altered inflammatory signaling, extracellular matrix turnover, oxidative stress responses, and tissue repair. This narrative review summarizes current knowledge of the structural, immunological, vascular, and epigenetic features of gingival aging, with particular emphasis on the relationship among biological aging, epigenetic regulation, and periodontal disease susceptibility. The review also highlights the implications of gingival aging for frailty, oral-systemic health, and future preventive or therapeutic strategies. Because gingiva-specific longitudinal human evidence remains limited, epigenetic biomarkers and aging-targeted interventions should be regarded as promising yet investigational approaches requiring further validation.
REFERENCE [16] · ID: 42412415
ID: 42412415 Title: Intercellular mitochondrial transfer and trans-mitophagy in response to protein import dysfunction. Abstract: Mitochondrial protein import is critical for organelle biogenesis, maintenance, and regeneration-essential for cellular homeostasis. Import dysfunction compromises cellular energy supplies, which is damaging to cells, particularly those with high energetic demands like neurons. Previously, we have shown that import failure is rescued by intercellular mitochondrial transfer (IMT) via tunnelling nanotubes (TNTs) however, the fate of the transferred mitochondria and the mechanistic basis for rescue were unresolved. Here, we show that bidirectional mitochondrial trafficking between cells harboring import-defective and import-competent mitochondria is distinct in terms of their regulation and ensuing consequences. Transferred import-defective mitochondria are highly fragmented and destined for canonical lysosomal degradation. In contrast, reactive oxygen species (ROS)-producing mitochondria at the periphery of cells with import-competent mitochondria are transferred into neighboring cells undergoing import failure. These new arrivals then accumulate within previously uncharacterized "mitochondrial degradation bodies" (MDBs). We speculate that the cooperation of these distinct cases of TNT-mediated conventional and noncanonical "trans-mitophagy" instigates mitochondrial regeneration, and thereby rescues mitochondrial function.